Содержание
-
Русское название
-
Английское название
-
Латинское название
-
Брутто формула
-
Фармакологическая группа вещества Сарилумаб
-
Нозологическая классификация
-
Код CAS
-
Фармакологическое действие
-
Характеристика
-
Фармакология
-
Применение вещества Сарилумаб
-
Противопоказания
-
Ограничения к применению
-
Применение при беременности и кормлении грудью
-
Побочные действия вещества Сарилумаб
-
Взаимодействие
-
Передозировка
-
Способ применения и дозы
-
Меры предосторожности
-
Источники информации
-
Торговые названия с действующим веществом Сарилумаб
Русское название
Сарилумаб
Английское название
Sarilumab
Латинское название
Sarilumabum (род. Sarilumabi)
Брутто формула
C6388H9918N1718O1998S44
Фармакологическая группа вещества Сарилумаб
Нозологическая классификация
Код CAS
1189541-98-7
Фармакологическое действие
—
иммунодепрессивное.
Характеристика
Человеческое моноклональное антитело (подтип IgG1) к рецептору ИЛ-6.
Фармакология
Фармакодинамика
Механизм действия
Сарилумаб специфически связывается как с растворимыми, так и с мембранными рецепторами ИЛ-6 (IL-6Rα), и подавляет ИЛ-6-опосредованную передачу сигнала с вовлечением убиквитарного сигнального белка гликопротеина 130 (gp130) и STAT-3 белков (трансдукторы сигналов и активаторы транскрипции-3).
В функциональных исследованиях на человеческих клетках было показано, что сарилумаб способен блокировать сигнальный путь ИЛ-6, измеряемый по степени подавления STAT-3 белков, только в присутствии ИЛ-6. ИЛ-6 — это плейотропный цитокин, который стимулирует различные клеточные ответы, такие как пролиферация, дифференцировка, выживаемость и апоптоз клеток; активирует высвобождение белков острой фазы воспаления в гепатоцитах, включая С-реактивный белок (CPБ) и сывороточный амилоид А. Повышенный уровень ИЛ-6, выявляемый в синовиальной жидкости у пациентов с ревматоидным артритом, играет важную роль как в развитии патологического воспаления, так и деструкции суставов, которые являются отличительными признаками ревматоидного артрита. ИЛ-6 участвует в различных физиологических процессах, таких как миграция и активация Т- и В-лимфоцитов, моноцитов и остеокластов, приводя к развитию системного воспаления, воспаления синовиальной оболочки суставов и костных эрозий у пациентов с ревматоидным артритом. Действие сарилумаба приводит к уменьшению воспаления и сопровождается изменениями лабораторных показателей, такими как снижение абсолютного числа нейтрофилов (АЧН) и повышение концентрации липидов.
Фармакодинамические эффекты
После п/к введения сарилумаба в разовой дозе 150 и 200 мг у пациентов с ревматоидным артритом наблюдалось быстрое снижение уровня CPБ. Уровень CPБ снижался до нормальных значений уже через 4 дня после начала лечения. У пациентов с ревматоидным артритом после введения разовой дозы сарилумаба АЧН снижалось до минимального значения в течение 3–4 дней, а затем восстанавливалось до исходного уровня. Лечение сарилумабом приводило к снижению уровня фибриногена и сывороточного амилоида А, а также к повышению уровня Hb и альбумина сыворотки крови.
Клиническая эффективность и безопасность
Эффективность и безопасность сарилумаба были оценены в трех рандомизированных двойных слепых контролируемых многоцентровых исследованиях.
Плацебо-контролируемые исследования
В исследовании MOBILITY принимали участие 1197 пациентов с ревматоидным артритом с недостаточным клиническим ответом на терапию метотрексатом. Пациенты получали сарилумаб в дозе 200, 150 мг или плацебо каждые 2 нед одновременно с метотрексатом. В исследовании TARGET принимали участие 546 пациентов с ревматоидным артритом с недостаточным клиническим ответом на терапию одним или несколькими антагонистами ФНО-α или в случае их непереносимости. Пациенты получали сарилумаб в дозе 200 или 150 мг или плацебо в сочетании с традиционными болезньмодифицирующими антиревматическими препаратами (БМАРП) каждые 2 нед.
Клинический ответ
На 24-й нед терапии в обоих исследованиях у пациентов, получавших сарилумаб в дозе 200 или 150 мг в сочетании с традиционными БМАРП 1 раз каждые 2 нед, частота ответа ACR20, ACR50 и ACR70 была выше, чем у пациентов, получавших плацебо. В открытой продленной фазе исследования эти результаты сохранялись в течение 3 лет терапии.
В исследовании MOBILITY к 52-й нед бóльшая часть пациентов, получавших сарилумаб в дозе 200 или 150 мг 1 раз каждые 2 нед в сочетании с метотрексатом, достигла ремиссии, определяемой по DAS28-CPБ (индекс активности болезни по 28 суставам — СРБ) <2,6 по сравнению с группой пациентов, получавших плацебо в сочетании с метотрексатом.
В исследованиях MOBILITY и TARGET в группе активного лечения более высокая частота ответа по критериям ACR20 по сравнению с группой плацебо наблюдалась уже через 2 нед, результаты сохранялись на протяжении всего исследования.
Результаты исследования MOBILITY через 52 нед терапии были сходны с результатами исследования TARGET через 24 нед.
Рентгенологический ответ
В исследовании MOBILITY эффективность обеих доз сарилумаба в сочетании с метотрексатом превосходила эффективность комбинации плацебо и метотрексата в отношении структурных повреждений суставов, оцениваемых по изменению модифицированного счета Шарпа/ван дер Хейде, по сравнению с исходным уровнем через 24 и 52 нед. Через 52 нед терапии сарилумабом в дозе 200 и 150 мг в сочетании с метотрексатом было отмечено уменьшение прогрессирования структурных повреждений на 91 и 68% соответственно по сравнению с комбинацией плацебо и метотрексата.
Изменения функционального статуса
В исследованиях MOBILITY и TARGET к 16-й и 12-й нед неделе терапии соответственно было продемонстрировано более выраженное улучшение функционального статуса по HAQ-DI в группах пациентов, получавших сарилумаб, по сравнению с группами плацебо, которое сохранялось до 52-й нед в исследовании MOBILITY.
Исследование с использованием активного препарата в качестве контроля
Исследование MONARCH—24-недельное рандомизированное двойное слепое, двойное маскированное исследование, в котором сравнивали монотерапию сарилумабом в дозе 200 мг с монотерапией адалимумабом в дозе 40 мг.
Сарилумаб в дозе 200 мг превосходил адалимумаб в дозе 40 мг в отношении снижения активности заболевания и улучшения функционального статуса.
Фармакокинетика
Фармакокинетика сарилумаба исследовалась у 2186 пациентов с ревматоидным артритом, из которых 751 пациент получал его в дозе 150 мг и 891 пациент — в дозе 200 мг п/к 1 раз каждые 2 нед в течение до 52 нед. Медиана Cmax достигалась через 2–4 дня после введения сарилумаба.
Всасывание
В равновесном состоянии концентрация сарилумаба в интервалах между введениями, которая измерялась с помощью AUC, увеличивалась в 2 раза при повышении дозы со 150 до 200 мг при введении 1 раз каждые 2 нед. Равновесное состояние достигалось через 12–16 нед с 2–3-кратным накоплением по сравнению с концентрацией после введения разовой дозы.
При введении дозы 150 мг 1 раз каждые 2 нед расчетные средние значения (±стандартное отклонение) в равновесном состоянии AUC, Cmin и Сmах сарилумаба составили (210±115) мг·сут/л, (6,95±7,6) мг/л и (20,4±8,27) мг/л, соответственно. При введении дозы 200 мг 1 раз каждые 2 нед расчетные средние значения (±стандартное отклонение) в равновесном состоянии AUC, Cmin и Сmах сарилумаба составили (396±194) мг·сут/л, (16,7±13,5) и (35,4±13,9) мг/л соответственно.
Распределение
У пациентов с ревматоидным артритом кажущийся Vss составил 8,3 л.
Метаболизм
Метаболизм сарилумаба не изучен. Предполагается, что сарилумаб, как и другие моноклональные антитела, распадается на небольшие пептиды и аминокислоты через катаболизм таким же образом, как и эндогенный IgG.
Выведение
Выведение сарилумаба происходит одновременно двумя путями: линейным и нелинейным. При высоких концентрациях выведение осуществляется преимущественно посредством линейного ненасыщаемого протеолитического пути, в то время как при более низких концентрациях преобладает нелинейное, насыщаемое, опосредованное мишенями выведение. Эти параллельные пути определяют начальный T1/2 от 8 до 10 дней и терминальный T1/2, зависящий от концентрации, от 2 до 4 дней.
После достижения равновесного состояния при введении последней дозы сарилумаба 150 и 200 мг медиана времени до неопределяемых концентраций составляет 30 и 49 дней соответственно. Моноклональные антитела не выводятся почками и печенью.
Линейность
У пациентов с ревматоидным артритом наблюдалась более чем дозозависимое увеличение фармакокинетической экспозиции. В равновесном состоянии концентрация в перерывах между введениями сарилумаба измерялась AUC, которая увеличивалась примерно в 2 раза с повышением дозы в 1,33 раза, от 150 до 200 мг, при введении 1 раз каждые 2 нед.
Взаимодействие с субстратами цитохрома Р450
Симвастатин является субстратом изофермента CYP3A4 и транспортного белка ОАТР1В1. У 17 пациентов с ревматоидным артритом через неделю после разового п/к введения сарилумаба в дозе 200 мг экспозиция симвастатина и симвастатиновой кислоты уменьшилась на 45 и 36% соответственно.
Особые группы пациентов
Возраст, пол, этническая принадлежность и масса тела. Популяционный анализ фармакокинетики у взрослых пациентов с ревматоидным артритом (возраст от 18 до 88 лет; 14% пациентов в возрасте старше 65 лет) показал, что возраст, пол и этническая принадлежность не оказывают значимого влияния на фармакокинетику сарилумаба. У пациентов с массой тела более 100 кг применение сарилумаба в дозах 150 и 200 мг продемонстрировало эффективность, однако пациенты с массой тела более 100 кг получили бóльшую терапевтическую пользу при применении дозы 200 мг.
Нарушение функции почек. Официальных исследований влияния почечной недостаточности на фармакокинетику сарилумаба не проводилось. Почечная недостаточность легкой и средней степени тяжести не влияет на фармакокинетику сарилумаба. Пациентам с почечной недостаточностью легкой и средней степени тяжести коррекция дозы не требуется. Применение сарилумаба у пациентов с почечной недостаточностью тяжелой степени не изучалось.
Нарушение функции печени. Официальных исследований влияния печеночной недостаточности на фармакокинетику сарилумаба не проводилось.
Применение вещества Сарилумаб
Лечение ревматоидного артрита умеренной или высокой степени активности у взрослых пациентов в сочетании с метотрексатом при недостаточном ответе на терапию одним или несколькими БМАРП или при их непереносимости (в монотерапии при непереносимости или нецелесообразности терапии метотрексатом).
Противопоказания
Повышенная чувствительность к сарилумабу; активные серьезные инфекционные заболевания (см. «Меры предосторожности»); дети в возрасте до 18 лет (эффективность и безопасность у детей с ревматоидным артритом не установлены).
Ограничения к применению
Хроническая или рецидивирующая инфекция; серьезные или оппортунистические инфекции в анамнезе; сопутствующие заболевания, предрасполагающие к развитию инфекций; после контакта с больными туберкулезом; проживание или посещение регионов, эндемичных по туберкулезу или микозам (необходимо оценить соотношение пользы и риска перед началом применения, см. «Меры предосторожности»); ВИЧ-инфекция; повышенный риск развития перфорации ЖКТ (см. «Меры предосторожности»); пожилой возраст (в связи с более высокой частотой развития инфекций, см. «Меры предосторожности»).
Применение при беременности и кормлении грудью
Женщины детородного возраста должны использовать эффективные методы контрацепции во время терапии сарилумабом и в течение 3 мес после ее окончания.
Данные по применению сарилумаба у беременных женщин ограничены или отсутствуют. Известно, что моноклональные антитела проникают через плаценту, при этом большее количество антител проникает через плаценту в III триместре.
Исследования на животных не дают прямых или косвенных указаний на негативное влияние сарилумаба с точки зрения репродуктивной токсичности. Сарилумаб не следует применять во время беременности за исключением тех случаев, когда потенциальная польза применения для матери превышает потенциальный риск для плода.
Неизвестно, экскретируется ли сарилумаб в грудное молоко или подвергается системной абсорбции у новорожденного после грудного вскармливания. Информация относительно влияния сарилумаба на ребенка, находящегося на грудном вскармливании, или продукцию грудного молока отсутствует. Поскольку IgG1 в небольших количествах могут экскретироваться в грудное молоко, следует с учетом пользы грудного вскармливания для ребенка и пользы терапии для женщины принять решение либо о прекращении грудного вскармливания, либо об отмене сарилумаба.
Фертильность
Данные о воздействии сарилумаба на фертильность у людей отсутствуют. Исследования на животных показали отсутствие негативного влияния на фертильность самок и самцов.
Побочные действия вещества Сарилумаб
Наиболее частыми нежелательными реакциями, которые наблюдались в клинических исследованиях, были нейтропения, повышение активности АЛТ, эритема в месте инъекции, инфекции верхних отделов дыхательных путей, инфекции мочевыводящих путей.
Наиболее частыми серьезными нежелательными реакциями были инфекции.
Безопасность сарилумаба в сочетании с БМАРП оценивалась на основании данных 7 клинических исследований, 2 из которых были плацебо-контролируемыми, с включением 2887 пациентов (выборка для оценки долгосрочной безопасности). Из них 2170 пациентов получали сарилумаб не менее 24 нед, 1546 пациентов — не менее 48 нед, 1020 — не менее 96 нед и 624 — не менее 144 нед.
Частота нежелательных реакций, перечисленных ниже, определялась следующим образом: очень часто (≥1/10); часто (≥1/100, <1/10); нечасто (≥1/1000, <1/100); редко (≥1/10000, <1/1000); очень редко (<1/10000). В пределах каждой частотной группы нежелательные реакции представлены в порядке уменьшения серьезности.
Инфекционные и паразитарные заболевания: часто — инфекции верхних отделов дыхательных путей, инфекции мочевыводящих путей, назофарингит, герпес ротовой полости.
Со стороны крови и лимфатической системы: очень часто — нейтропения; часто — тромбоцитопения.
Со стороны печени и желчевыводящих путей: часто — повышение активности печеночных трансаминаз.
Со стороны обмена веществ и питания: часто — гипертриглицеридемия, гиперхолестеринемия.
Общие расстройства и реакции в месте введения: часто — эритема и зуд в месте введения.
Описание отдельных нежелательных реакций
Инфекции. В популяции пациентов, участвовавших в плацебо-контролируемых исследованиях, распространенность инфекций составила 84,5; 81 и 75,1 случая на 100 пациенто-лет для комбинации сарилумаба в дозе 200 мг и БМАРП, сарилумаба в дозе 150 мг и БМАРП и плацебо и БМАРП соответственно. Наиболее частыми инфекциями (от 5 до 7% пациентов) были инфекции верхних отделов дыхательных путей, инфекции мочевыводящих путей и назофарингит. Частота серьезных инфекций составила 4,3; 3 и 3,1 случая на 100 пациенто-лет для комбинации сарилумаба в дозе 200 мг и БМАРП, сарилумаба в дозе 150 мг и БМАРП и плацебо и БМАРП соответственно.
При оценке долгосрочной безопасности в популяции пациентов, получавших сарилумаб в сочетании с БМАРП, частота инфекций и серьезных инфекций составила 57,3 и 3,4 случая на 100 пациенто-лет соответственно. Наиболее частыми серьезными инфекциями были пневмония и целлюлит (воспаление подкожной жировой клетчатки). Были зарегистрированы случаи оппортунистических инфекций.
Общая частота инфекций и серьезных инфекций в популяции пациентов, получавших сарилумаб в виде монотерапии, была сопоставима с частотой в популяции пациентов, получавших терапию сарилумабом в сочетании с БМАРП.
Перфорация ЖКТ. В популяции пациентов, участвовавших в плацебо-контролируемых исследованиях, у одного пациента, получавшего сарилумаб, развилась перфорация ЖКТ (0,11 случая на 100 пациенто-лет). При оценке долгосрочной безопасности в популяции пациентов, получавших сарилумаб в сочетании с БМАРП, частота перфораций ЖКТ составила 0,14 случая на 100 пациенто-лет.
Сообщения о перфорации ЖКТ в основном регистрировали как осложнения дивертикулита, включая перфорацию нижних отделов ЖКТ и абсцесс. Большинство пациентов с развившейся перфорацией ЖКТ получали сопутствующую терапию НПВС, ГКС или метотрексатом. Неизвестно, как дополнительно влияют эти ЛС на развитие перфорации ЖКТ при одновременном применении с сарилумабом.
В популяции пациентов, получавших сарилумаб в монотерапии, о перфорациях ЖКТ не сообщалось.
Реакции гиперчувствительности. В популяции пациентов, участвовавших в плацебо-контролируемых исследованиях, доля пациентов, которые прекратили лечение из-за реакций гиперчувствительности, была выше среди пациентов, получавших сарилумаб (0,9% — в группе пациентов, получавших сарилумаб в дозе 200 мг, 0,5% — в группе пациентов, получавших сарилумаб в дозе 150 мг), чем в группе плацебо (0,2%).
При оценке долгосрочной безопасности частота отмены сарилумаба из-за реакций гиперчувствительности в популяции пациентов, получавших его в сочетании с БМАРП, и в популяции пациентов, получавших сарилумаб в виде монотерапии, была сопоставима с частотой в популяции пациентов из плацебо-контролируемых исследований.
В плацебо-контролируемых исследованиях серьезные нежелательные реакции гиперчувствительности развились у 0,2% пациентов, которые получали сарилумаб в дозе 200 мг каждые 2 нед в сочетании с БМАРП, и ни одного случая не было отмечено в группе пациентов, получавших сарилумаб в дозе 150 мг каждые 2 нед в сочетании с БМАРП.
Реакции в месте введения. В популяции пациентов, участвовавших в плацебо-контролируемых исследованиях, реакции в месте введения сарилумаба были зарегистрированы у 9,5, 8 и 1,4% пациентов, получавших сарилумаб в дозе 200, 150 мг и плацебо соответственно. У большинства пациентов реакции в месте введения (включая эритему и зуд) были легкой степени тяжести. В связи с реакциями в месте введения сарилумаб был преждевременно отменен у двух пациентов (0,2%).
Отклонения лабораторных показателей.
Для того чтобы обеспечить прямое сравнение частоты отклонений лабораторных показателей между группами плацебо и активного лечения, были использованы данные, полученные за период 0–12 нед, поскольку они были получены до того, как пациентов можно было перевести с плацебо на сарилумаб.
Количество нейтрофилов. Снижение количества нейтрофилов <1×109/л отмечалось у 6,4 и 3,6% пациентов в группах, получавших сарилумаб в дозе 200 мг в сочетании с БМАРП и сарилумаб в дозе 150 мг в сочетании с БМАРП соответственно; в группе плацебо в сочетании с БМАРП данная нежелательная реакция не наблюдалась. Снижение количества нейтрофилов <0,5×109/л отмечалось у 0,8 и 0,6% пациентов в группах, получавших сарилумаб в дозе 200 мг в сочетании с БМАРП и сарилумаб в дозе 150 мг в сочетании с БМАРП соответственно. У пациентов со снижением АЧН изменение схемы лечения, например прерывание терапии сарилумабом или снижение дозы, приводило к увеличению или нормализации АЧН. Снижение АЧН не сопровождалось более высокой частотой развития инфекций, включая серьезные.
При оценке долгосрочной безопасности в популяции пациентов, получавших сарилумаб в сочетании с БМАРП, и в популяции пациентов, получавших монотерапию сарилумабом, наблюдения относительно числа нейтрофилов были сопоставимы с наблюдениями, полученными для популяции пациентов из плацебо-контролируемых исследований.
Количество тромбоцитов. Снижение количества тромбоцитов <100×103/мкл наблюдалось у 1,2 и 0,6% пациентов в группах, получавших сарилумаб в дозе 200 мг в сочетании с БМАРП и сарилумаб в дозе 150 мг в сочетании с БМАРП соответственно; в группе пациентов, получавших плацебо в сочетании с БМАРП, данная нежелательная реакция не наблюдалась.
При оценке долгосрочной безопасности в популяции пациентов, получавших сарилумаб в сочетании с БМАРП, и в популяции пациентов, получавших монотерапию сарилумабом, наблюдения относительно количества тромбоцитов были сопоставимы с наблюдениями, полученными для популяции пациентов из плацебо-контролируемых исследований.
Кровотечений, связанных со снижением количества тромбоцитов, зарегистрировано не было.
Ферменты печени. Изменения показателей печеночных ферментов представлены в таблице ниже. У пациентов с повышением активности печеночных трансаминаз изменение схемы лечения, т.е прерывание терапии сарилумабом или снижение дозы, приводило к снижению или нормализации активности печеночных трансаминаз. Эти изменения не сопровождались ни клинически значимым повышением концентрации прямого билирубина, ни клиническими проявлениями гепатита или печеночной недостаточности.
Таблица
Частота повышения активности печеночных трансаминаз в контролируемых клинических исследованиях, %
| Показатель | Плацебо + БМАРП (n=661) | Сарилумаб 150 мг + БМАРП (n=660) | Сарилумаб 200 мг + БМАПР (n=661) | Сарилумаб (монотерапия, любая доза) (n=467) |
| АСТ | ||||
| >3,5×ВГН | 0 | 1,2 | 1,1 | 1,1 |
| >5×ВГН | 0 | 0,6 | 0,2 | 0 |
| АЛТ | ||||
| >3–5×ВГН | 0,6 | 3,2 | 2,4 | 1,9 |
| >5×ВГН | 0 | 1,1 | 0,8 | 0,2 |
Липиды. В популяции пациентов, участвовавших в плацебо-контролируемых исследованиях, параметры липидного профиля (ЛПНП, ЛПВП и триглицериды) впервые оценивались через 4 нед после начала лечения комбинацией сарилумаба и БМАРП. На 4-й нед терапии среднее значение ЛПНП увеличилось на 14 мг/дл, триглицеридов — на 23 мг/дл, ЛПВП — на 3 мг/дл. После 4-й нед терапии дополнительного повышения этих показателей не наблюдалось. Значимых различий между дозами отмечено не было.
При оценке долгосрочной безопасности в популяции пациентов, получавших сарилумаб в сочетании с БМАРП, и в популяции пациентов, получавших сарилумаб в монотерапии, данные показателей липидного профиля были сопоставимы с наблюдениями, полученными для популяции пациентов из плацебо-контролируемых исследований.
Иммуногенностъ. Как и все белковые ЛС, сарилумаб обладает потенциалом иммуногенности. В популяции пациентов, участвовавших в плацебо-контролируемых исследованиях, у 4, 5,6 и 2% пациентов, получавших сарилумаб в дозе 200 мг в сочетании с БМАРП, сарилумаб в дозе 150 мг в сочетании с БМАРП и комбинацию плацебо и БМАРП соответственно, выявлена положительная реакция на антитела к сарилумабу. Положительная реакция на нейтрализующие антитела обнаружена у 1, 1,6 и 0,2% пациентов, получавших сарилумаб в дозе 200, 150 мг и плацебо соответственно.
Данные в популяции пациентов, получавших сарилумаб в монотерапии, были сопоставимы с результатами в популяции пациентов, получавших сарилумаб в сочетании с БМАРП.
Образование антител к сарилумабу может повлиять на его фармакокинетику. Корреляции между образованием антител к сарилумабу и потерей эффективности терапии или развитием нежелательных реакций не наблюдалось.
Определение иммунного ответа во многом зависит от чувствительности и специфичности используемых методов, способа и времени забора образцов, сопутствующей терапии и основного заболевания. По этим причинам сравнение частоты выработки антител к сарилумабу с частотой выработки антител к другим ЛС может быть недостоверным.
Злокачествениые новообразования. В популяции пациентов, участвовавших в плацебо-контролируемых исследованиях, частота развития злокачественных новообразований у пациентов, получавших либо сарилумаб в сочетании с БМАРП, либо комбинацию плацебо и БМАРП, была одинаковой (1 случай на 100 пациенто-лет).
При оценке долгосрочной безопасности в популяции пациентов, получавших сарилумаб в сочетании с БМАРП, и в популяции пациентов, получавших сарилумаб в монотерапии, наблюдения относительно частоты злокачественных новообразований были сопоставимы с полученными в популяции пациентов из плацебо-контролируемых исследований.
Взаимодействие
Применение с другими ЛС для лечения ревматоидного артрита
Одновременное применение с метотрексатом не влияло на экспозицию сарилумаба. Не ожидается также влияния сарилумаба на экспозицию метотрексата при их одновременном применении, клинические данные отсутствуют.
Одновременное применение сарилумаба с ингибиторами янус-киназы (ингибиторы JAK) или другими биологическими БМАРП, такими как антагонисты ФНО, антагонисты рецептора ИЛ-1R, анти-СD20 моноклональные антитела, селективные ко-стимулирующие модуляторы, не изучалось. Следует избегать одновременного применения сарилумаба с биологическими БМАРП.
Взаимодействие с ЛС, являющимися субстратами цитохрома Р450
Различные исследования in vitro и ограниченное количество исследований in vivo у человека показали, что цитокины и модуляторы цитокинов могут влиять на экспрессию и активность специфических изоферментов цитохрома P450 (CYP1A2, CYP2C19, CYP3A4) и, таким образом, имеют возможность изменять фармакокинетику одновременно принимаемых ЛС, являющихся субстратами для этих изоферментов. Повышение концентрации ИЛ-6 может снижать активность цитохрома Р450 у пациентов с ревматоидным артритом и, следовательно, повышать у них концентрацию ЛС, являющихся субстратами цитохрома Р450, по сравнению с пациентами без ревматоидного артрита. Блокада сигнального пути ИЛ-6 антагонистами рецепторов ИЛ-6Rα, такими как сарилумаб, может устранить ингибирующее действие ИЛ-6 и восстановить активность цитохрома Р450, приводя к изменению концентрации ЛС.
Изменение влияния ИЛ-6 на изоферменты цитохрома Р450 под действием сарилумаба может быть клинически значимо для субстратов цитохрома Р450 с узким терапевтическим диапазоном концентрации, для которых доза корректируется индивидуально. После начала применения или отмены сарилумаба у пациентов, получающих лечение ЛС, являющимися субстратами цитохрома Р450, следует проводить мониторинг терапевтического эффекта (например, для варфарина) или концентрации ЛС (например, для теофиллина) и корректировать дозу ЛС по мере необходимости.
Следует соблюдать осторожность при начале терапии сарилумабом у пациентов, принимающих ЛС, которые являются субстратами изофермента CYP3A4 (например, пероральные контрацептивы или статины), т.к. сарилумаб может устранить ингибирующий эффект ИЛ-6 и восстанавливать активность изофермента CYP3A4, приводя к снижению экспозиции и активности субстратов CYP3A4. Взаимодействие сарилумаба с субстратами других изоферментов цитохрома P450 (CYPP2C9, CYP2C19, CYP2D6) не изучалось.
Передозировка
Имеются ограниченные данные по передозировке сарилумаба. Специфического лечения при передозировке сарилумаба не существует. В случае передозировки следует тщательно контролировать состояние пациента, проводить симптоматическую и поддерживающую терапию.
Способ применения и дозы
П/к, 1 раз каждые 2 нед.
Меры предосторожности
Серьезные инфекции
В период лечения сарилумабом следует тщательно контролировать пациентов на предмет развития симптомов и признаков инфекций. Поскольку среди пациентов пожилого возраста частота развития инфекций выше, следует с осторожностью проводить лечение данной категории пациентов.
Сарилумаб не следует применять у пациентов с активным течением инфекционного заболевания, включая локализованные инфекции.
Необходимо оценить соотношение пользы и риска перед началом применения сарилумаба у пациентов с хронической или рецидивирующей инфекцией, серьезными или оппортунистическими инфекциями в анамнезе, ВИЧ-инфекцией, сопутствующими заболеваниями, предрасполагающими к развитию инфекций, после контакта с больными туберкулезом, проживавших или посещавших регионы, эндемичные по туберкулезу или микозам.
Следует прервать лечение сарилумабом, если у пациента отмечается развитие серьезной или оппортунистической инфекции.
Пациент, у которого в период лечения сарулимабом развилась инфекция, должен незамедлительно пройти полное диагностическое обследование, предусмотренное для лиц с ослабленным иммунитетом, затем ему должна быть назначена адекватная антибактериальная терапия с последующим тщательным наблюдением.
У пациентов, получавших иммунодепрессивные ЛС для лечения ревматоидного артрита, включая сарилумаб, были зарегистрированы серьезные инфекции, иногда с летальным исходом, вызванные бактериальными, микобактериальными, инвазивными грибковыми, вирусными и другими оппортунистическими патогенами. Наиболее часто наблюдавшимися серьезными инфекциями при применении сарилумаба были пневмония и целлюлит (воспаление подкожной жировой клетчатки). Из оппортунистических инфекций при применении сарилумаба были зарегистрированы туберкулез, кандидоз и пневмоцистоз. В единичных случаях наблюдались диссеминированные, а не локализованные инфекции у пациентов, часто получающих сопутствующую терапию иммунодепрессивными ЛС, такими как метотрексат или ГКС, что в сочетании с ревматоидным артритом может предрасполагать к развитию инфекции.
Туберкулез
До начала лечения сарилумабом у пациентов необходимо оценить наличие факторов риска развития туберкулеза и провести обследование на латентную инфекцию. Пациентам с латентным или активным туберкулезом следует до начала лечения сарилумабом провести стандартную противотуберкулезную терапию. У пациентов с латентным или активным туберкулезом в анамнезе, у которых невозможно подтвердить, проводился ли необходимый курс терапии, и у пациентов с отрицательным результатом анализа на латентный туберкулез, но имеющих факторы риска развития туберкулезной инфекции, следует рассмотреть возможность проведения противотуберкулезной терапии до начала лечения сарилумабом. При решении вопроса о проведении противотуберкулезной терапии целесообразно проконсультироваться с фтизиатром.
Следует тщательно контролировать пациентов на предмет развития признаков и симптомов туберкулеза, включая пациентов, чей результат обследования на латентный туберкулез до начала терапии был отрицательным.
Реактивация вирусной инфекции
При применении иммунодепрессивных биологических препаратов сообщалось о реактивации вирусных инфекций. В клинических исследованиях сарилумаба отмечались случаи опоясывающего герпеса. В клинических исследованиях случаи реактивации вируса гепатита В зарегистрированы не были, однако из исследований были исключены пациенты, имеющие риск реактивации инфекции.
Лабораторные показатели
Количество нейтрофилов. При лечении сарилумабом отмечалась более высокая частота снижения АЧН. Снижение АЧН не сопровождалось более высокой частотой развития инфекций, включая серьезные инфекции. Не рекомендуется начинать лечение сарилумабом у пациентов с АЧН <2×109/л. У пациентов со снижением АЧН <0,5×109/л лечение сарилумабом следует отменить.
Следует контролировать число нейтрофилов через 4–8 нед после начала терапии сарилумабом и далее — в зависимости от клинических показаний.
При изменении дозы сарилумаба следует ориентироваться на показатели, полученные в конце интервала между введениями.
Количество тромбоцитов. В клинических исследованиях при лечении сарилумабом отмечалось снижение количества тромбоцитов. Снижение количества тромбоцитов не сопровождалось развитием кровотечения.
Не рекомендуется начинать лечение сарилумабом у пациентов с количеством тромбоцитов <150×103/мкл. При снижении количества тромбоцитов <50×103/мкл терапию сарилумабом следует отменить.
Следует контролировать количество тромбоцитов через 4–8 нед после начала терапии и далее — в зависимости от клинических показаний.
Ферменты печени. При лечении сарилумабом отмечалась более высокая частота повышения активности печеночных ферментов, что в клинических исследованиях носило транзиторный характер и не приводило к появлению каких-либо клинически выраженных симптомов повреждения печени. Увеличение частоты и выраженности повышения активности печеночных ферментов наблюдалось при применении сарилумаба в сочетании с потенциально гепатотоксичными ЛС (например, метотрексат).
Не рекомендуется начинать лечение сарилумабом у пациентов с повышением активности печеночных трансаминаз АЛТ или ACT >1,5×ВГН. При повышении активности АЛТ >5×ВГН терапию сарилумабом следует отменить.
Следует контролировать активность АЛТ и ACT через 4–8 нед после начала терапии и далее — каждые 3 мес. В случае клинической необходимости следует рассмотреть возможность исследования других показателей функции печени, таких как билирубин.
Изменение показателей липидного обмена. У пациентов с хроническими воспалительными заболеваниями концентрация липидов в крови может быть снижена. Лечение сарилумабом сопровождалось повышением концентрации липидов, таких как Хс ЛПНП, Хс ЛПВП и/или триглицериды.
Необходимо контролировать показатели липидного обмена приблизительно через 4–8 нед после начала терапии сарилумабом, затем — примерно через каждые 6 мес.
Лечение пациентов осуществляется согласно клиническим руководствам по ведению пациентов с гиперлипидемией.
Перфорация ЖКТ. В клинических исследованиях было зарегистрировано такое нежелательное явление, как перфорация ЖКТ, которая, прежде всего, является осложнением дивертикулита. Следует с осторожностью применять сарилумаб у пациентов, имеющих в анамнезе указания на язвы ЖКТ или дивертикулит. Необходимо незамедлительно обращать внимание на появление у пациентов новых абдоминальных симптомов, таких как стойкая боль и повышение температуры.
Злокачественные новообразования. Лечение иммунодепрессивными ЛС может приводить к увеличению риска развития злокачественных новообразований. Влияние терапии сарилумабом на развитие злокачественных новообразований неизвестно, но в клинических исследованиях были зарегистрированы случаи злокачественных новообразований.
Реакции гиперчувствительности. Сообщалось о развитии реакций гиперчувствительности, связанных с применением сарилумаба. Наиболее частыми реакциями гиперчувствительности были сыпь в месте введения, кожная сыпь и крапивница. Пациент должен быть проинформирован о необходимости незамедлительного обращения к врачу в случае появления любых реакций гиперчувствительности. При развитии анафилактических реакций или реакций гиперчувствительности введение сарилумаба прекратить немедленно. Сарилумаб не назначают пациентам с известной гиперчувствительностью к нему.
Нарушение функции печени. Пациентам с активными заболеваниями печени или нарушениями функции печени лечение сарилумабом не рекомендуется.
Вакцинация. Следует избегать одновременного применения живых, а также живых аттенуированных вакцин во время лечения сарилумабом, поскольку клиническая безопасность данного взаимодействия не установлена. Отсутствуют данные о вторичной передаче возбудителей заболеваний от лиц, вакцинированных живыми вакцинами, пациентам, получающим сарилумаб. Перед началом лечения сарилумабом рекомендуется, чтобы все пациенты были вакцинированы в соответствии с действующими рекомендациями по вакцинации. Интервал между вакцинацией живыми вакцинами и началом лечения сарилумабом должен соответствовать действующим рекомендациям по вакцинации в отношении одновременного применения иммунодепрессивных ЛС.
Кардиоваскулярный риск. Пациенты с ревматоидным артритом имеют повышенный риск развития сердечно-сосудистых заболеваний. Факторы риска (например, артериальная гипертензия, гиперлипидемия) необходимо учитывать в рамках проведения стандартной терапии.
Дети. Не следует применять сарилумаб у детей в возрасте до 18 лет (в настоящий момент безопасность и эффективность не установлены).
Влияние на способность управлять транспортными средствами и механизмами. Сарилумаб не оказывает или оказывает незначительное влияние на способность управлять транспортными средствами или работать с механизмами.
Источники информации
Обобщенные материалы www.grls.rosminzdrav.ru, 2018.
Торговые названия с действующим веществом Сарилумаб
| Торговое название | Цена за упаковку, руб. |
|---|---|
| Кевзара® |
64964.00 |
Фармакологическое действие
Сарилумаб — человеческое моноклональное антитело (подтип IgG1) к рецептору интерлейкина-6 (ИЛ-6). Специфически связывается как с растворимыми, так и с мембранными рецепторами ИЛ-6 (IL-6Rα), подавляет ИЛ-6-опосредованную передачу сигнала с вовлечением убиквитарного сигнального белка гликопротеина 130 (gp130) и STAT-3 белков (трансдукторы сигналов и активаторы транскрипции-3). Сарилумаб способен блокировать сигнальный путь ИЛ-6, измеряемый по степени подавления STAT-3 белков, только в присутствии ИЛ-6.
ИЛ-6 участвует в различных физиологических процессах, таких как миграция и активация Т- и В-лимфоцитов, моноцитов и остеокластов, приводя к развитию системного воспаления, воспаления синовиальной оболочки суставов и развитию костных эрозий у пациентов с ревматоидным артритом. Действие сарилумаба приводит к уменьшению воспаления и сопровождается изменениями лабораторных показателей, такими как снижение абсолютного числа нейтрофилов и повышение концентрации липидов.
Фармакокинетика
При введении дозы 150 мг 1 раз каждые 2 недели расчетные средние значения в равновесном состоянии AUC, Cmin и Сmax сарилумаба составили 210±115 мг×сут/л, 6.95±7.6 мг/л и 20.4±8.27 мг/л соответственно. При введении дозы 200 мг 1 раз каждые 2 недели расчетные средние значения в равновесном состоянии AUC, Cmin и Сmax сарилумаба составили 396±194 мг×сут/л, 16.7±13.5 мг/л и 35.4±13.9 мг/л соответственно.
В равновесном состоянии концентрация сарилумаба в интервалах между введениями увеличивалась в 2 раза при повышении дозы со 150 мг до 200 мг при введении 1 раз каждые 2 недели. Равновесное состояние достигалось через 12-16 недель с 2-3-кратным накоплением по сравнению с концентрацией после введения разовой дозы.
У пациентов с ревматоидным артритом кажущийся Vd в равновесном состоянии составил 8.3 л. Наблюдалось более чем дозозависимое увеличение фармакокинетической экспозиции. В равновесном состоянии концентрация в перерывах между введениями препарата измерялась AUC, которая увеличивалась примерно в 2 раза с повышением дозы в 1.33 раза от 150 до 200 мг при введении препарата 1 раз каждые 2 недели.
Предполагается, что сарилумаб, как и другие моноклональные антитела, распадается на небольшие пептиды и аминокислоты через катаболизм, таким же образом, как и эндогенный иммуноглобулин (IgG).
Выведение сарилумаба происходит одновременно двумя путями: линейным и нелинейным. При высоких концентрациях выведение осуществляется преимущественно посредством линейного ненасыщаемого протеолитического пути, в то время как при более низких концентрациях преобладает нелинейное, насыщаемое, опосредованное мишенями, выведение. Эти параллельные пути определяют начальный Т1/2 от 8 до 10 дней и терминальный Т1/2, зависящий от концентрации, от 2 до 4 дней. После достижения равновесного состояния при введении последней дозы сарилумаба 150 мг и 200 мг медиана времени до неопределяемых концентраций, составляет 30 и 49 дней соответственно. Моноклональные антитела не выводятся почками и печенью.
Показания активного вещества
САРИЛУМАБ
В комбинации с метотрексатом для лечения ревматоидного артрита умеренной или высокой степени активности у взрослых при недостаточном ответе на терапию одним или несколькими болезнь-модифицирующими антиревматическими препаратами или при их непереносимости; в качестве монотерапии при непереносимости метотрексата или при нецелесообразности терапии метотрексатом.
Режим дозирования
Препарат вводят п/к. Рекомендуемая доза составляет 200 мг 1 раз каждые 2 недели.
При развитии нейтропении, тромбоцитопении, повышении активности печеночных ферментов рекомендуется уменьшить дозу до 150 мг 1 раз каждые 2 недели.
Побочное действие
Инфекционные и паразитарные заболевания: часто — инфекции верхних отделов дыхательных путей, инфекции мочевыводящих путей, назофарингит, герпес ротовой полости.
Со стороны системы кроветворения: очень часто — нейтропения; часто — тромбоцитопения.
Со стороны печени и желчевыводящих путей: повышение активности печеночных трансаминаз.
Со стороны обмена веществ: гипертриглицеридемия, гиперхолестеринемия.
Прочие: часто — эритема и зуд в месте введения препарата.
Противопоказания к применению
Повышенная чувствительность к сарилумабу; детский и подростковый возраст до 18 лет.
Применение при беременности и кормлении грудью
Сарилумаб не следует применять во время беременности, за исключением тех случаев, когда потенциальная польза применения для матери превышает потенциальный риск для плода.
Женщинам детородного возраста необходимо использовать эффективные методы контрацепции во время терапии сарилумабом и в течение 3 месяцев после ее окончания.
Т.к. IgG1 в небольших количествах могут выделяться с грудным молоком, следует с учетом пользы грудного вскармливания для ребенка и необходимости терапии для женщины принять решение либо о прекращении грудного вскармливания, либо об отмене сарилумаба.
Применение при нарушениях функции печени
Пациентам с активными заболеваниями печени или нарушениями функции печени лечение сарилумабом не рекомендуется.
Применение при нарушениях функции почек
У пациентов с почечной недостаточностью легкой и средней степени тяжести коррекции дозы препарата не требуется.
Применение у детей
Противопоказано применение в возрасте до 18 лет.
Применение у пожилых пациентов
С осторожностью следует назначать сарилумаб пациентам пожилого возраста.
Особые указания
С осторожностью следует назначать сарилумаб пациентам с хронической или рецидивирующей инфекцией; серьезными или оппортунистическими инфекциями в анамнезе; сопутствующими заболеваниями, предрасполагающими к развитию инфекций; после контакта с больными туберкулезом; проживавших или посещавших регионы, эндемичные по туберкулезу или микозам; пациентам с ВИЧ-инфекцией; пациентам с повышенным риском развития перфорации ЖКТ; пациентам пожилого возраста.
Лекарственное взаимодействие
Следует избегать одновременного применения сарилумаба с биологическими болезнь-модифицирующими антиревматическими препаратами.
После начала применения или отмены сарилумаба пациентам, получающим лечение лекарственными препаратами, являющимися субстратами цитохрома Р450, следует проводить мониторинг терапевтического эффекта (например, для варфарина) или концентрации лекарственного препарата (например, для теофиллина) и корректировать дозу препарата по мере необходимости.
Следует соблюдать осторожность в начале терапии сарилумабом у пациентов, принимающих препараты, которые являются субстратами изофермента CYP3A4 (например, пероральные контрацептивные средства или статины), т.к. сарилумаб может устранить ингибирующий эффект ИЛ-6 и восстановить активность изофермента CYP3A4, приводя к снижению экспозиции и активности субстратов CYP3A4.
Серьезные инфекции
В период лечения препаратом Кевзара® следует тщательно контролировать пациентов на предмет развития симптомов и признаков инфекций. Поскольку среди пациентов пожилого возраста частота развития инфекций выше, следует с осторожностью проводить лечение данной категории пациентов.
Препарат Кевзара® не следует применять у пациентов с активным течением инфекционного заболевания, включая локализованные инфекции. Необходимо оценить соотношение пользы и риска перед началом применения препарата Кевзара® у пациентов:
- с хронической или рецидивирующей инфекцией;
- с серьезными или оппортунистическими инфекциямив анамнезе;
- с ВИЧ-инфекцией;
- с сопутствующими заболеваниями, предрасполагающими к развитию инфекций;
- после контакта с больными туберкулезом;
- проживавших или посещавших регионы, эндемичные по туберкулезу или микозам.
Следует прервать лечение препаратом Кевзара®, если у пациента отмечается развитие серьезной или оппортунистической инфекции.
Пациент, у которого в период лечения препаратом Кевзара® развилась инфекция, должен незамедлительно пройти полное диагностическое обследование, предусмотренное для лиц с ослабленным иммунитетом; затем ему должна быть назначена адекватная антибактериальная терапия с последующим тщательным наблюдением.
У пациентов, получавших иммунодепрессивные препараты для лечения ревматоидного артрита, включая препарат Кевзара®, были зарегистрированы серьезные инфекции, иногда с летальным исходом, вызванные бактериальными, микобактериальными, инвазивными грибковыми, вирусными и другими оппортунистическими патогенами. Наиболее часто наблюдавшимися серьезными инфекциями при применении препарата Кевзара® были пневмония и целлюлит (воспаление подкожной жировой клетчатки). Из оппортунистических инфекций при применении препарата Кевзара® были зарегистрированы туберкулез, кандидоз и пневмоцистоз. В единичных случаях наблюдались диссеминированные, а не локализованные инфекции у пациентов, часто получающих сопутствующую терапию иммунодепрессивными препаратами, такими как метотрексат или глюкокортикостероиды, что в сочетании с ревматоидным артритом, может предрасполагать к развитию инфекции.
Туберкулез
До начала лечения препаратом Кевзара® у пациентов необходимо оценить наличие факторов риска туберкулеза и провести обследование на латентную инфекцию. Пациентам с латентным или активным туберкулезом следует до начала лечения препаратом Кевзара® провести стандартную противотуберкулезную терапию. У пациентов с латентным или активным туберкулезом в анамнезе, у которых невозможно подтвердить, проводился ли необходимый курс терапии, и у пациентов с отрицательным результатом анализа на латентный туберкулез, но имеющих факторы риска развития туберкулезной инфекции, следует рассмотреть возможность проведения противотуберкулезной терапии до начала лечения препаратом Кевзара®. При решении вопроса о проведении противотуберкулезной терапии целесообразно проконсультироваться с фтизиатром.
Следует тщательно контролировать пациентов на предмет развития признаков и симптомов туберкулеза, включая пациентов, чей результат обследования на латентный туберкулез до начала терапии был отрицательным.
Реактивация вирусной инфекции
При применении иммунодепрессивных биологических препаратов сообщалось о реактивации вирусных инфекций. В клинических исследованиях препарата Кевзара® отмечались случаи опоясывающего герпеса. В клинических исследованиях случаи реактивации вируса гепатита В зарегистрированы не были, однако из исследований были исключены пациенты, имеющие риск реактивации инфекции.
Лабораторные показатели
Количество нейтрофилов
При лечении препаратом Кевзара® отмечалась более высокая частота снижения АЧН. Снижение АЧН не сопровождалось более высокой частотой развития инфекций, включая серьезные инфекции.
Не рекомендуется начинать лечение препаратом Кевзара® у пациентов с АЧН <2×109/л. У пациентов со снижением АЧН <0,5×109/л лечение препаратом Кевзара следует отменить.
Следует контролировать число нейтрофилов через 4-8 недель после начала терапии препаратом Кевзара® и далее – в зависимости от клинических показаний. Рекомендации по изменению дозы на основании значений АЧН представлены в разделе «Способ применения и дозы».
При изменении дозы препарата Кевзара® следует ориентироваться на показатели, полученные в конце интервала между введениями препарата.
Количество тромбоцитов
В клинических исследованиях при лечении препаратом Кевзара® отмечалось снижение количества тромбоцитов. Снижение количества тромбоцитов не сопровождалось развитием кровотечения.
Не рекомендуется начинать лечение препаратом Кевзара® у пациентов с количеством тромбоцитов <150×103/мкл. При снижении количества тромбоцитов <50×103/мкл терапию препаратом Кевзара® следует отменить. Следует контролировать количество тромбоцитов через 4-8 недель после начала терапии и далее – в зависимости от клинических показаний. Рекомендации по изменению дозы препарата на основании количества тромбоцитов представлены в разделе «Способ применения и дозы».
Ферменты печени
При лечении препаратом Кевзара® отмечалась более высокая частота повышения активности «печеночных» ферментов, что в клинических исследованиях носило транзиторный характер и не приводило к появлению каких-либо клинически выраженных симптомов повреждения печени. Увеличение частоты и выраженности повышения активности «печеночных» ферментов наблюдалось при применении препарата Кевзара® в комбинации с потенциально гепатотоксичными препаратами (например, метотрексатом).
Не рекомендуется начинать лечение препаратом Кевзара® у пациентов с повышением активности «печеночных» трансаминаз аланинаминотрансферазы (АЛТ) или аспартатаминотрансферазы (ACT) >1,5×ВГН. При повышении активности АЛТ >5×ВГН терапию препаратом Кевзара® следует отменить.
Следует контролировать активность АЛТ и ACT через 4-8 недель после начала терапии и далее – каждые 3 месяца. В случае клинической необходимости следует рассмотреть возможность исследования других показателей функции печени, таких как билирубин. Рекомендации по изменению дозы на основании повышения активности «печеночных» трансаминаз представлены в разделе «Способ применения и дозы».
Изменение показателей липидного обмена
У пациентов с хроническими воспалительными заболеваниями концентрации липидов в крови могут быть снижены. Лечение препаратом Кевзара® сопровождалось повышением концентраций липидов, таких как холестерин ЛПНП, холестерин ЛПВП и/или триглицериды.
Необходимо контролировать показатели липидного обмена приблизительно через 4-8 недель после начала терапии препаратом Кевзара®, затем – примерно через каждые 6 месяцев.
Лечение пациентов осуществляется согласно клиническим руководствам по ведению пациентов с гиперлипидемией.
Перфорация желудочно-кишечного тракта
В клинических исследованиях было зарегистрировано такое нежелательное явление, как перфорация желудочно-кишечного тракта, которая, прежде всего, является осложнением дивертикулита. Следует с осторожностью применять препарат Кевзара® у пациентов, имеющих в анамнезе указания на язвы желудочно-кишечного тракта или дивертикулит. Необходимо незамедлительно обращать внимание на появление у пациентов новых абдоминальных симптомов, таких как стойкая боль и повышение температуры.
Злокачественные новообразования
Лечение иммунодепрессивными препаратами может приводить к увеличению риска развития злокачественных новообразований. Влияние терапии препаратом Кевзара® на развитие злокачественных новообразований неизвестно, но в клинических исследованиях были зарегистрированы случаи злокачественных новообразований.
Реакции гиперчувствительности
Сообщалось о развитии реакций гиперчувствительности, связанных с приемом препарата Кевзара®. Наиболее частыми реакциями гиперчувствительности были сыпь в месте введения препарата, кожная сыпь и крапивница. Пациента следует информировать о незамедлительном обращении к врачу в случае появления любых реакций гиперчувствительности. При развитии анафилактических реакций или реакций гиперчувствительности введение препарата Кевзара® следует прекратить немедленно. Препарат Кевзара® не назначают пациентам с известной гиперчувствительностью к сарилумабу.
Нарушение функции печени
Пациентам с активными заболеваниями печени или нарушениями функции печени лечение препаратом Кевзара® не рекомендуется.
Вакцинация
Следует избегать одновременного применения живых, а также живых аттенуированных вакцин во время лечения препаратом Кевзара®, поскольку клиническая безопасность данного взаимодействия не установлена. Отсутствуют данные о вторичной передаче возбудителей заболеваний от лиц, вакцинированных живыми вакцинами, пациентам, получающим препарат Кевзара®. Перед началом лечения препаратом Кевзара® рекомендуется, чтобы все пациенты были вакцинированы в соответствии с действующими рекомендациями по вакцинации. Интервал между вакцинацией живыми вакцинами и началом лечения препаратом Кевзара® должен соответствовать действующим рекомендациям по вакцинации в отношении одновременного применения иммунодепрессивных препаратов.
Кардиоваскулярный риск
Пациенты с ревматоидным артритом имеют повышенный риск развития сердечно-сосудистых заболеваний. Факторы риска (например, артериальная гипертензия, гиперлипидемия) необходимо учитывать в рамках проведения стандартной терапии.
Применение у детей
Не следует применять препарат Кевзара® у детей в возрасте до 18 лет (в настоящий момент безопасность и эффективность препарата не установлены).
Несовместимость
Ввиду отсутствия исследований на совместимость, препарат Кевзара® нельзя смешивать с другими лекарственными средствами.
Кевзара — инструкция по применению
Синонимы, аналоги
Статьи
Регистрационный номер:
ЛП-005185
Торговое наименование препарата:
Кевзара®
Международное непатентованное наименование:
сарилумаб
Лекарственная форма:
раствор для подкожного введения.
Состав
| Ингредиент | Количество (дозировка 150 мг) |
Количество (дозировка 200 мг) |
||
| в 1 мл | в шприце/шприц-ручке | в 1 мл | в шприце/шприц-ручке | |
| Действующее вещество | ||||
| Сарилумаб | 131,6 мг | 150 мг | 175 мг | 200 мг |
| Вспомогательные вещества | ||||
| L-гистидин L-гистидина гидрохлорида моногидрат |
3,26 мг1 | 3,71 мг1 | 3,26 мг1 | 3,71 мг1 |
| L-аргинина гидрохлорид |
7,84 мг2 | 8,94 мг2 | 7,84 мг2 | 8,94 мг2 |
| Сахароза | 50 мг | 57 мг | 50 мг | 57 мг |
| Полисорбат-20 | 2 мг | 2,28 мг | 2 мг | 2,28 мг |
| Вода для инъекций | до 1,00 мл | до 1,14 мл | до 1,00 мл | до 1,14 мл |
1) Содержание L-гистидина и L-гистидина гидрохлорида моногидрата, приведено в пересчете на L-гистидин.
2) Содержание L-аргинина гидрохлорида приведено в пересчете на L-аргинин.
Описание
Прозрачная или опалесцирующая, бесцветная или коричневато-желтого цвета жидкость.
Фармакотерапевтическая группа:
иммунодепрессанты, ингибиторы интерлейкина.
Код ATX:
L04AC14.
Фармакологические свойства
Фармакодинамика
Механизм действия
Сарилумаб – человеческое моноклональное антитело (подтип IgGl) к рецептору интерлейкина-6 (ИЛ-6). Сарилумаб специфически связывается как с растворимыми, так и с мембранными рецепторами ИЛ-6 (IL-6Rα), и подавляет ИЛ-6-опосредованную передачу сигнала с вовлечением убиквитарного сигнального белка гликопротеина 130 (gp130) и STAT-3 белков (трансдукторы сигналов и активаторы транскрипции-3).
В функциональных исследованиях на человеческих клетках было показано, что сарилумаб способен блокировать сигнальный путь ИЛ-6, измеряемый по степени подавления STAT-3 белков, только в присутствии ИЛ-6.
ИЛ-6 – это плейотропный цитокин, который стимулирует различные клеточные ответы, такие как пролиферация, дифференцировка, выживаемость и апоптоз клеток; активирует высвобождение белков острой фазы воспаления в гепатоцитах, включая С-реактивный белок (СРБ) и сывороточный амилоид А. Повышенный уровень ИЛ-6, выявляемый в синовиальной жидкости у пациентов с ревматоидным артритом, играет важную роль как в развитии патологического воспаления, так и в развитии деструкции суставов, которые являются отличительными признаками ревматоидного артрита. ИЛ-6 участвует в различных физиологических процессах, таких как миграция и активация Т- и В-лимфоцитов, моноцитов и остеокластов, приводя к развитию системного воспаления, воспаления синовиальной оболочки суставов и развитию костных эрозий у пациентов с ревматоидным артритом. Действие сарилумаба приводит к уменьшению воспаления и сопровождается изменениями лабораторных показателей, такими как снижение абсолютного числа нейтрофилов (АЧН) и повышение концентрации липидов.
Фармакодинамические эффекты
После подкожного введения сарилумаба в разовых дозах 150 мг и 200 мг у пациентов с ревматоидным артритом наблюдалось быстрое снижение уровня СРБ. Уровень СРБ снижался до нормальных значений уже через 4 дня после начала лечения. У пациентов с ревматоидным артритом после введения разовой дозы сарилумаба АЧН снижалось до минимального значения в течение 3-4 дней, а затем восстанавливалось до исходного уровня. Лечение сарилумабом приводило к снижению уровня фибриногена и сывороточного амилоида А, а также к повышению уровней гемоглобина и альбумина сыворотки крови.
Клиническая эффективность и безопасность
Эффективность и безопасность препарата Кевзара® были оценены в 3-х рандомизированных двойных слепых контролируемых многоцентровых исследованиях.
Плацебо-контролируемые исследования
В исследовании MOBILITY принимали участие 1197 пациентов с ревматоидным артритом с недостаточным клиническим ответом на терапию метотрексатом. Пациенты получали препарат Кевзара® в дозах 200 мг, 150 мг или плацебо каждые 2 недели одновременно с метотрексатом. В исследовании TARGET принимали участие 546 пациентов с ревматоидным артритом с недостаточным клиническим ответом на терапию одним или несколькими антагонистами ФНО-α или в случае их непереносимости. Пациенты получали препарат Кевзара® в дозах 200 мг или 150 мг или плацебо в сочетании с традиционными болезнь-модифицирующими антиревматическими препаратами [тБМАРП] каждые 2 недели.
• Клинический ответ
На 24-й неделе терапии в обоих исследованиях у пациентов, получавших препарат Кевзара® в дозе 200 мг или 150 мг в сочетании с тБМАРП 1 раз каждые 2 недели частота ответа ACR20, ACR50 и ACR70 была выше, чем у пациентов, получавших плацебо. В открытой продленной фазе исследования эти результаты сохранялись в течение 3-х лет терапии.
В исследовании MOBILITY к 52-й неделе большая часть пациентов, получавших препарат Кевзара в дозе 200 мг или 150 мг 1 раз каждые 2 недели в сочетании с метотрексатом, достигла ремиссии, определяемой по DAS28-CPБ<2,6 (Индексу активности болезни по 28 суставам – С-реактивный белок), по сравнению с группой пациентов, получавших плацебо в сочетании с метотрексатом.
В исследованиях MOBILITY и TARGET в группе активного лечения более высокая частота ответа по критериям ACR20 по сравнению с группой плацебо наблюдалась уже через 2 недели; результаты сохранялись на протяжении всего исследования.
Результаты исследования MOBILITY через 52 недели терапии были сходны с результатами исследования TARGET через 24 недели.
• Рентгенологический ответ
В исследовании MOBILITY эффективность обеих доз препарата Кевзара® в сочетании с метотрексатом превосходила эффективность комбинации плацебо и метотрексата в отношении структурных повреждений суставов, оцениваемых по изменению модифицированного счёта Шарпа/ван дер Хейде по сравнению с исходным уровнем через 24 и через 52 недели.
Через 52 недели терапии препаратом Кевзара® в дозе 200 мг и в дозе 150 мг в сочетании с метотрексатом было отмечено уменьшение прогрессирования структурных повреждений на 91% и 68%, соответственно, по сравнению с комбинацией плацебо и метотрексата.
• Изменения функционального статуса
В исследованиях MOBILITY и TARGET к 16-й и 12-й неделе неделе терапии соответственно было продемонстрировано более выраженное улучшение функционального статуса по HAQ-DI в группах препарата Кевзара® по сравнению с плацебо, которое сохранялось до 52 недели в исследовании MOBILITY.
Исследование с использованием активного препарата в качестве контроля
Исследование MONARCH – 24-недельное рандомизированное двойное слепое, двойное маскированное исследование, в котором сравнивали монотерапию препаратом Кевзара® в дозе 200 мг с монотерапией адалимумабом в дозе 40 мг.
Препарат Кевзара® в дозе 200 мг превосходил адалимумаб в дозе 40 мг в отношении снижения активности заболевания и улучшения функционального статуса.
Фармакокинетика
Фармакокинетика сарилумаба исследовалась у 2186 пациентов с ревматоидным артритом, из которых 751 пациент получал препарат Кевзара® в дозе 150 мг и 891 пациент – в дозе 200 мг подкожно 1 раз каждые 2 недели в течение до 52-х недель. Медиана максимальной концентрации достигалась через 2-4 дня после введения препарата.
Всасывание
В равновесном состоянии концентрация сарилумаба в интервалах между введениями, которая измерялась с помощью площади под кривой зависимости концентрации от времени (AUC), увеличивалась в 2 раза при повышении дозы со 150 мг до 200 мг при введении 1 раз каждые 2 недели. Равновесное состояние достигалось через 12-16 недель с 2-3-кратным накоплением по сравнению с концентрацией после введения разовой дозы.
При введении дозы 150 мг 1 раз каждые 2 недели расчетные средние значения (±стандартное отклонение) в равновесном состоянии AUC, минимальная концентрация (Cmin) и максимальная концентрация (Сmax) сарилумаба составили 210±115 мг·сут/л, 6,95±7,6 мг/л и 20,4±8,27 мг/л, соответственно.
При введении дозы 200 мг 1 раз каждые 2 недели расчетные средние значения (±стандартное отклонение) в равновесном состоянии AUC, Cmin и Сmax сарилумаба составили 396±194 мг·сут/л, 16,7±13,5 мг/л и 35,4±13,9 мг/л, соответственно.
Распределение
У пациентов с ревматоидным артритом кажущийся объем распределения в равновесном состоянии составил 8,3 л.
Метаболизм
Метаболизм сарилумаба не изучен. Предполагается, что сарилумаб, как и другие моноклональные антитела, распадается на небольшие пептиды и аминокислоты через катаболизм таким же образом, как и эндогенный иммуноглобулин (IgG).
Выведение
Выведение сарилумаба происходит одновременно двумя путями: линейным и нелинейным. При высоких концентрациях выведение осуществляется преимущественно посредством линейного ненасыщаемого протеолитического пути, в то время как при более низких концентрациях преобладает нелинейное, насыщаемое, опосредованное мишенями, выведение. Эти параллельные пути определяют начальный период полувыведения от 8 до 10 дней и терминальный период полувыведения, зависящий от концентрации, от 2 до 4 дней.
После достижения равновесного состояния при введении последней дозы сарилумаба 150 мг и 200 мг медиана времени до неопределяемых концентраций, составляет 30 и 49 дней, соответственно. Моноклональные антитела не выводятся почками и печенью.
Линейность/нелинейность
У пациентов с ревматоидным артритом наблюдалась более чем дозозависимое увеличение фармакокинетической экспозиции. В равновесном состоянии концентрация в перерывах между введениями препарата измерялась AUC, которая увеличивалась примерно в 2 раза с повышением дозы в 1,33 раза от 150 до 200 мг при введении препарата 1 раз каждые 2 недели.
Взаимодействие с субстратами цитохрома Р450
Симвастатин является субстратом изофермента CYP3A4 и транспортного белка ОАТР1В1. У 17 пациентов с ревматоидным артритом через неделю после разового подкожного введения сарилумаба в дозе 200 мг, экспозиция симвастатина и симвастатиновой кислоты уменьшилась на 45% и 36% соответственно.
Особые группы пациентов
Возраст, пол, этническая принадлежность и масса тела
Популяционный анализ фармакокинетики у взрослых пациентов с ревматоидным артритом (возраст от 18 до 88 лет; 14% пациентов в возрасте старше 65 лет), показал, что возраст, пол и этническая принадлежность не оказывают значимого влияния на фармакокинетику сарилумаба. У пациентов с массой тела более 100 кг применение сарилумаба в обеих дозах 150 мг и 200 мг продемонстрировало эффективность; однако пациенты с массой тела более 100 кг получили большую терапевтическую пользу при применении дозы 200 мг.
Нарушения функции почек
Каких-либо официальных исследований влияния почечной недостаточности на фармакокинетику сарилумаба не проводилось. Почечная недостаточность легкой и средней степени тяжести не влияет на фармакокинетику сарилумаба. Пациентам с почечной недостаточностью легкой и средней степени тяжести коррекция дозы не требуется. Применение сарилумаба у пациентов с почечной недостаточностью тяжелой степени не изучалось.
Нарушения функции печени
Каких-либо официальных исследований влияния печеночной недостаточности на фармакокинетику сарилумаба не проводилось.
Показания к применению
Препарат Кевзара® в комбинации с метотрексатом показан для лечения ревматоидного артрита умеренной или высокой степени активности у взрослых пациентов при недостаточном ответе на терапию одним или несколькими болезнь-модифицирующими антиревматическими препаратами (БМАРП) или при их непереносимости.
Препарат Кевзара® может назначаться в монотерапии при непереносимости метотрексата или при нецелесообразности терапии метотрексатом.
Противопоказания
- Повышенная чувствительность к активному веществу или любому вспомогательному компоненту препарата.
- Активные серьезные инфекционные заболевания (см. раздел «Особые указания»).
- Дети в возрасте до 18 лет в связи с неустановленными эффективностью и безопасностью у детей с ревматоидным артритом.
С осторожностью
- У пациентов с хронической или рецидивирующей инфекцией; серьезными или оппортунистическими инфекциями в анамнезе; с сопутствующими заболеваниями, предрасполагающими к развитию инфекций; после контакта с больными туберкулезом; проживавших или посещавших регионы, эндемичные по туберкулезу или микозам (необходимо оценить соотношение пользы и риска перед началом применения, см. раздел «Особые указания»).
- У пациентов с ВИЧ-инфекцией;
- У пациентов с повышенным риском развития перфорации желудочно-кишечного тракта (см. раздел «Особые указания»).
- У пациентов пожилого возраста (в связи с более высокой частотой развития инфекций у данной категории пациентов, см. раздел «Особые указания»).
- Ограничения по применению препарата в зависимости от возраста пациента приведены в разделе «Способ применения и дозы».
Применение при беременности и период грудного вскармливания
Женщины детородного возраста
Женщины детородного возраста должны использовать эффективные методы контрацепции во время терапии препаратом Кевзара® и в течение 3-х месяцев после ее окончания.
Беременность
Данные по применению сарилумаба у беременных женщин ограничены или отсутствуют. Известно, что моноклональные антитела проникают через плаценту, при этом большее количество антител проникает через плаценту в III триместре.
Исследования на животных не дают прямых или косвенных указаний на негативное влияние сарилумаба с точки зрения репродуктивной токсичности. Препарат Кевзара® не следует применять во время беременности, за исключением тех случаев, когда потенциальная польза применения для матери превышает потенциальный риск для плода.
Период грудного вскармливания
Неизвестно, экскретируется ли сарилумаб в грудное молоко или подвергается ли системной абсорбции у новорожденного после грудного вскармливания. Информация относительно влияния сарилумаба на ребенка, находящегося на грудном вскармливании, или продукцию грудного молока отсутствует. Поскольку IgGl в небольших количествах могут экскретироваться в грудное молоко, следует с учетом пользы грудного вскармливания для ребенка и пользы терапии для женщины принять решение либо о прекращении грудного вскармливания, либо об отмене сарилумаба.
Фертильность
Данные о воздействии сарилумаба на фертильность у людей отсутствуют. Исследования на животных показали отсутствие негативного влияния на фертильность самок и самцов.
Способ применения и дозы
Лечение препаратом Кевзара® должно назначаться и проводиться под контролем специалистов, имеющих опыт диагностики и лечения ревматоидного артрита.
Рекомендуемая доза препарата Кевзара® составляет 200 мг 1 раз каждые 2 недели. Препарат вводят подкожно.
При развитии нейтропении, тромбоцитопении, повышении активности «печеночных» ферментов рекомендуется уменьшить дозу с 200 мг 1 раз каждые 2 недели до 150 мг 1 раз каждые 2 недели.
Коррекция дозы
При развитии серьезных инфекций следует прекратить лечение препаратом Кевзара® до установления контроля над инфекционным процессом.
Не рекомендуется начинать лечение препаратом Кевзара® у пациентов со снижением абсолютного числа нейтрофилов (АЧН) менее чем 2×109/л.
Не рекомендуется начинать лечение препаратом Кевзара® у пациентов со снижением количества тромбоцитов ниже 150×103/мкл.
Рекомендации по коррекции дозы при развитии нейтропении, тромбоцитопении или при повышении активности «печеночных» ферментов приведены ниже в таблицах.
Таблица 1. Низкое значение абсолютного числа нейтрофилов.
| Значение показателя (число клеток×109/л) |
Рекомендации |
| АЧН >1 | Назначенную дозу препарата Кевзара® не изменяют. |
| АЧН 0.5-1 | Следует прервать лечение препаратом Кевзара® до восстановления АЧН >1×109/л. Затем можно возобновить лечение в дозе 150 мг 1 раз каждые 2 недели и увеличить до 200 мг 1 раз каждые 2 недели в соответствии с клинической необходимостью. |
| АЧН <0,5 | Следует отменить препарат Кевзара®. |
Таблица 2. Снижение количества тромбоцитов.
| Значение показателя (число клеток×103/мкл) |
Рекомендации |
| 50-100 | Следует прервать лечение препаратом Кевзара® до восстановления количества тромбоцитов >100×103/мкл. Затем можно возобновить лечение препаратом Кевзара® в дозе 150 мг 1 раз каждые 2 недели и увеличить до 200 мг 1 раз каждые 2 недели в соответствии с клинической необходимостью. |
| <50 | Если при повторном анализе крови количество тромбоцитов <50×103/мкл, следует отменить препарат Кевзара®. |
Таблица 3. Повышение активности «печеночных» ферментов.
| Значение активности аланинаминотрансферазы (АЛТ) | Рекомендации |
| Превышение ВГН* в 1-3 раза | При клинической необходимости следует рассмотреть коррекцию дозы одновременно принимаемых БМАРП. |
| Превышение ВГН в 3-5 раз | Следует прервать лечение препаратом Кевзара® до снижения активности АЛТ до уровня, менее чем в 3 раза превышающего ВГН. Затем можно возобновить лечение препаратом Кевзара® в дозе 150 мг 1 раз каждые 2 недели и увеличить до 200 мг 1 раз каждые 2 недели в соответствии с клинической необходимостью. |
| Превышение ВГН в 5 раз | Следует отменить препарат Кевзара®. |
| * – верхняя границы нормы. |
Пропуск дозы
Если введение препарата Кевзара® пропущено, и с момента пропуска введения препарата прошло 3 дня или менее, то следующую дозу необходимо ввести как можно скорее. Следующую очередную дозу вводят в обычное запланированное время.
Если с момента пропуска введения препарата прошло 4 дня или более, то следующую дозу вводят в следующее обычное запланированное время. При этом дозу нельзя удваивать.
Особые группы пациентов
Нарушения функции пачек
У пациентов с почечной недостаточностью легкой и средней степени тяжести коррекции дозы препарата не требуется. Применение препарата Кевзара® у пациентов с почечной недостаточностью тяжелой степени не изучалось.
Нарушения функции печени
Безопасность и эффективность препарата Кевзара® не изучались у пациентов с нарушениями функции печени, включая пациентов с положительными результатами серологических исследований на вирус гепатита В (HBV) или вирус гепатита С (HCV).
Пожилые пациенты
У пациентов в возрасте старше 65 лет коррекции дозы не требуется.
Дети
Не следует применять препарат Кевзара® у детей в возрасте до 18 лет (безопасность и эффективность препарата при ревматоидном артрите не установлены).
Способ применения
Препарат Кевзара® вводят подкожно.
Все содержимое (1,14 мл) предварительно заполненного шприца/предварительно заполненной шприц-ручки следует вводить подкожно. Места инъекций (область живота, наружная поверхность бедра, наружная поверхность плеча) следует чередовать при каждой инъекции. Не следует вводить препарат в болезненную и поврежденную кожу, в места с кровоподтеками и рубцами.
Пациент может самостоятельно выполнять подкожную инъекцию препарата Кевзара®, или ее может выполнять лицо, осуществляющее уход за пациентом. Пациент или лицо, осуществляющее уход за пациентом, до начала применения препарата Кевзара® должны быть обучены подготовке и введению препарата.
Побочное действие
Наиболее частыми нежелательными реакциями, которые наблюдались в клинических исследованиях, были нейтропения, повышение активности АЛТ, эритема в месте инъекции, инфекции верхних отделов дыхательных путей, инфекции мочевыводящих путей.
Наиболее частыми серьезными нежелательными реакциями были инфекции.
Безопасность препарата Кевзара® в сочетании с БМАРП оценивалась на основании данных 7 клинических исследований, 2 из которых были плацебо-контролируемыми, с включением 2887 пациентов (выборка для оценки долгосрочной безопасности). Из них 2170 пациентов получали препарат Кевзара® не менее 24 недель, 1546 пациентов – не менее 48 недель, 1020 пациентов – не менее 96 недель и 624 пациента – не менее 144 недель.
Частота нежелательных реакций, перечисленных ниже, определялась следующим образом: очень часто (≥1/10); часто (от ≥1/100 до <1/10); нечасто (от ≥1/1000 до <1/100); редко (от ≥1/10000 до <1/1000); очень редко (<1/10000). В пределах каждой частотной группы нежелательные реакции представлены в порядке уменьшения серьезности.
Инфекционные и паразитарные заболевания: часто – инфекции верхних отделов дыхательных путей, инфекции мочевыводящих путей, назофарингит, герпес ротовой полости.
Нарушения со стороны крови и лимфатической системы: очень часто – нейтропения; часто – тромбоцитопения.
Нарушения со стороны печени и желчевыводящих путей: часто – повышение активности «печеночных» трансаминаз.
Нарушения со стороны обмена веществ и питания: часто гипертриглицеридемия, гиперхолестеринемия.
Общие расстройства и реакции в месте введения: часто – эритема и зуд в месте введения препарата.
Описание отдельных нежелательных реакций
Инфекции
В популяции пациентов, участвующих в плацебо-контролируемых исследованиях, распространенность инфекций составила 84,5, 81,0 и 75,1 случаев на 100 пациенто-лет для комбинаций препарата Кевзара® в дозе 200 мг и БМАРП, препарата Кевзара® в дозе 150 мг и БМАРП и плацебо и БМАРП, соответственно. Наиболее частыми инфекциями (от 5% до 7% пациентов) были инфекции верхних отделов дыхательных путей, инфекции мочевыводящих путей и назофарингит. Частота серьезных инфекций составила 4,3, 3,0 и 3,1 случаев на 100 пациенто-лет для комбинаций препарата Кевзара® в дозе 200 мг и БМАРП, Кевзара® в дозе 150 мг и БМАРП и плацебо и БМАРП, соответственно.
При оценке долгосрочной безопасности в популяции пациентов, получавших препарат Кевзара® в сочетании с БМАРП, частота инфекций и серьезных инфекций составила 57,3 и 3,4 случаев на 100 пациенто-лет, соответственно. Наиболее частыми серьезными инфекциями были пневмонии и целлюлит (воспаление подкожной жировой клетчатки). Были зарегистрированы случаи оппортунистических инфекций.
Общая частота инфекций и серьезных инфекций в популяции пациентов, получавших препарат Кевзара® в виде монотерапии, была сопоставима с частотой в популяции пациентов, получавших терапию препаратом Кевзара® в сочетании с БМАРП.
Перфорация желудочно-кишечного тракта
В популяции пациентов, участвующих в плацебо-контролируемых исследованиях, у одного пациента, получавшего препарат Кевзара®, развилась перфорация желудочно-кишечного тракта (ЖКТ) (0,11 случая на 100 пациенто-лет). При оценке долгосрочной безопасности в популяции пациентов, получавших препарат Кевзара® в сочетании с БМАРП, частота перфораций ЖКТ составила 0,14 случая на 100 пациенто-лет.
Сообщения о перфорации ЖКТ в основном регистрировали как осложнения дивертикулита, включая перфорацию нижних отделов ЖКТ и абсцесс. Большинство пациентов с развившейся перфорацией ЖКТ получали сопутствующую терапию нестероидными противовоспалительными препаратами (НПВП), глюкокортикостероидами или метотрексатом. Неизвестно, как дополнительно влияют эти препараты на развитие перфорации ЖКТ при одновременном применении с препаратом Кевзара®.
В популяции пациентов, получавших препарат Кевзара® в монотерапии, о перфорациях ЖКТ не сообщалось.
Реакции гиперчувствительности
В популяции пациентов, участвующих в плацебо-контролируемых исследованиях, доля пациентов, которые прекратили лечение из-за реакций гиперчувствительности, была выше среди пациентов, получавших препарат Кевзара® (0,9% – в группе пациентов, получавших препарат в дозе 200 мг, 0,5% – в группе пациентов, получавших препарат в дозе 150 мг), чем в группе плацебо (0,2%).
При оценке долгосрочной безопасности частота отмены препарата Кевзара® из-за реакций гиперчувствительности в популяции пациентов, получавших препарат Кевзара® в сочетании с БМАРП, и в популяции пациентов, получавших препарат Кевзара® в виде монотерапии, была сопоставима с частотой в популяции пациентов из плацебо-контролируемых исследований.
В плацебо-контролируемых исследованиях серьезные нежелательные реакции гиперчувствительности развились у 0,2% пациентов, которые получали препарат Кевзара® в дозе 200 мг каждые 2 недели в сочетании с БМАРП, и ни одного случая не было отмечено в группе пациентов, получавших препарат Кевзара® в дозе 150 мг каждые 2 недели в сочетании с БМАРП.
Реакции в месте введения препарата
В популяции пациентов, участвующих в плацебо-контролируемых исследованиях, реакции в месте введения препарата были зарегистрированы у 9,5%, 8% и 1,4% пациентов, получавших препарат Кевзара® в дозах 200 мг, 150 мг и плацебо, соответственно. У большинства пациентов реакции в месте введения (включая эритему и зуд) были легкой степени тяжести. В связи с реакциями в месте введения препарат Кевзара® был преждевременно отменен у двух пациентов (0,2%).
Отклонения лабораторных показателей
Для того чтобы обеспечить прямое сравнение частоты отклонений лабораторных показателей между группами плацебо и активного лечения, были использованы данные, полученные за период 0-12 недель, поскольку они были получены до того, как пациентов можно было перевести с плацебо на препарат Кевзара®.
• Количество нейтрофилов
Снижение количества нейтрофилов <1×109/л отмечалось у 6,4% и 3,6% пациентов в группах, принимавших препарат Кевзара® в дозе 200 мг в сочетании с БМАРП и препарат Кевзара® в дозе 150 мг в сочетании с БМАРП, соответственно; в группе плацебо в сочетании с БМАРП данная нежелательная реакция не наблюдалась. Снижение количества нейтрофилов <0,5×109/л отмечалось у 0,8% и 0,6% пациентов в группах, принимавших препарат Кевзара® в дозе 200 мг в сочетании с БМАРП и препарат Кевзара® в дозе 150 мг в сочетании с БМАРП, соответственно. У пациентов со снижением АЧН изменение схемы лечения, например, прерывание терапии препаратом Кевзара® или снижение дозы, приводило к увеличению или нормализации АЧН. Снижение АЧН не сопровождалось более высокой частотой развития инфекций, включая серьезные инфекции.
При оценке долгосрочной безопасности в популяции пациентов, получавших препарат Кевзара® в сочетании с БМАРП, и в популяции пациентов, получавших монотерапию препаратом Кевзара®, наблюдения относительно числа нейтрофилов были сопоставимы с наблюдениями, полученными для популяции пациентов из плацебо-контролируемых исследований.
• Количество тромбоцитов
Снижение количества тромбоцитов <100×103/мкл наблюдалось у 1,2% и у 0,6% пациентов в группах, принимавших препарат Кевзара® в дозе 200 мг в сочетании с БМАРП и препарат Кевзара® в дозе 150 мг в сочетании с БМАРП, соответственно; в группе пациентов, получавших плацебо в сочетании с БМАРП, данная нежелательная реакция не наблюдалась.
При оценке долгосрочной безопасности в популяции пациентов, получавших препарат Кевзара® в сочетании с БМАРП, и в популяции пациентов, получавших монотерапию препаратом Кевзара®, наблюдения относительно количества тромбоцитов были сопоставимы с наблюдениями, полученными для популяции пациентов из плацебо-контролируемых исследований.
Кровотечений, связанных со снижением количества тромбоцитов, зарегистрировано не было.
• Ферменты печени
Изменения показателей «печеночных» ферментов представлены в Таблице 4.
У пациентов с повышением активности «печеночных» трансаминаз изменение схемы лечения, то есть прерывание терапии препаратом Кевзара® или снижение дозы, приводило к снижению или нормализации активности «печеночных» трансаминаз. Эти изменения не сопровождались ни клинически значимым повышением концентрации прямого билирубина, ни клиническими проявлениями гепатита или печеночной недостаточности.
Таблица 4. Частота повышения активности «печеночных» трансаминаз в контролируемых клинических исследованиях.
| Плацебо + БМАРП (n=661) |
Кевзара® 150 мг + БМАРП (n=660) |
Кевзара® 200 мг + БМАРП (n=661) |
Кевзара® (монотерапия, любая доза) (n=467) |
|
| ACT | ||||
| >3-5×ВГН | 0% | 1,2% | 1,1% | 1,1% |
| >5×ВГН | 0% | 0,6% | 0,2% | 0% |
| АЛТ | ||||
| >3-5×ВГН | 0,6% | 3,2% | 2,4% | 1,9% |
| >5×ВГН | 0% | 1,1% | 0,8% | 0,2% |
• Липиды
В популяции пациентов, участвующих в плацебо-контролируемых исследованиях, параметры липидного профиля (липопротеины низкой плотности [ЛПНП], липопротеины высокой плотности [ЛПВП] и триглицериды) впервые оценивались через 4 недели после начала лечения комбинацией препарата Кевзара® и БМАРП. На 4-й неделе терапии среднее значение ЛПНП увеличилось на 14 мг/дл, среднее значение триглицеридов – на 23 мг/дл, среднее значение ЛПВП – на 3 мг/дл. После 4-й недели терапии дополнительного повышения этих показателей не наблюдалось. Значимых различий между дозами отмечено не было.
При оценке долгосрочной безопасности в популяции пациентов, получавших препарат Кевзара® в сочетании БМАРП, и в популяции пациентов, получавших препарат Кевзара® в монотерапии, данные показателей липидного профиля были сопоставимы с наблюдениями, полученными для популяции пациентов из плацебо-контролируемых исследований.
Иммуногенностъ
Как и все белковые лекарственные препараты, препарат Кевзара® обладает потенциалом иммуногенности. В популяции пациентов, участвовавших в плацебо-контролируемых исследованиях, у 4%, 5,6% и 2% пациентов, получавших препарат Кевзара® в дозе 200 мг в сочетании с БМАРП, препарат Кевзара® в дозе 150 мг в сочетании с БМАРП и комбинацию плацебо и БМАРП, соответственно, выявлена положительная реакция на антитела к сарилумабу. Положительная реакция на нейтрализующие антитела обнаружена у 1%, 1,6% и 0,2% пациентов, получавших препарат Кевзара® в дозах 200 мг, 150 мг и плацебо, соответственно.
Данные в популяции пациентов, получавших препарат Кевзара® в монотерапии, были сопоставимы с результатами популяции пациентов, получавших препарат Кевзара® в сочетании с БМАРП.
Образование антител к сарилумабу может повлиять на его фармакокинетику. Корреляции между образованием антител к сарилумабу и потерей эффективности терапии или развитием нежелательных реакций не наблюдалось.
Определение иммунного ответа во многом зависит от чувствительности и специфичности используемых методов, способа и времени забора образцов, сопутствующей терапии и основного заболевания. По этим причинам сравнение частоты выработки антител к сарилумабу с частотой выработки антител к другим препаратам может быть недостоверным.
Злокачественные новообразования
В популяции пациентов, участвовавших в плацебо-контролируемых исследованиях, частота развития злокачественных новообразований у пациентов, получавших либо препарат Кевзара® в сочетании с БМАРП, либо комбинацию плацебо и БМАРП, была одинаковой (1,0 случай на 100 пациенто-лет).
При оценке долгосрочной безопасности в популяции пациентов, получавших препарат Кевзара® в сочетании с БМАРП, и в популяции пациентов, получавших препарат Кевзара® в монотерапии, наблюдения относительно частоты злокачественных новообразований были сопоставимы с наблюдениями, полученными в популяции пациентов из плацебо-контролируемых исследований.
Передозировка
Признаки и симптомы
Имеются ограниченные данные по передозировке препарат Кевзара®.
Лечение
Специфического лечения при передозировке препарата Кевзара® не существует. В случае передозировки следует тщательно контролировать состояние пациента, проводить симптоматическую и поддерживающую терапию.
Взаимодействие с другими лекарственными средствами
Применение с другими препаратами для лечения ревматоидного артрита
Одновременное применение с метотрексатом не влияло на экспозицию сарилумаба. Не ожидается также влияния сарилумаба на экспозицию метотрексата при их одновременном применении, клинические данные отсутствуют.
Одновременное применение препарата Кевзара® с ингибиторами янус-киназы (ингибиторами JAK) или другими биологическими БМАРП, такими как антагонисты ФНО, антагонисты рецептора интерлейкина-1 (ИЛ-1R), анти-CD20 моноклональные антитела, селективные ко-стимулирующие модуляторы, не изучалось. Следует избегать одновременного применения препарата Кевзара® с биологическими БМАРП.
Взаимодействие с препаратами, являющимися субстратами цитохрома Р450
Различные исследования in vitro и ограниченное количество исследований in vivo на человеке показали, что цитокины и модуляторы цитокинов могут влиять на экспрессию и активность специфических изоферментов цитохрома Р450 (CYP) (CYP1A2, CYP2C19, CYP3A4) и, таким образом, имеют возможность изменять фармакокинетику одновременно принимаемых препаратов, являющихся субстратами для этих изоферментов. Повышение концентрации ИЛ-6 может снижать активность цитохрома Р450 у пациентов с ревматоидным артритом, и, следовательно, повышать у них концентрацию препаратов, являющихся субстратами цитохрома Р450, по сравнению с пациентами без ревматоидного артрита. Блокада сигнального пути ИЛ-6 антагонистами рецепторов ИЛ-6Rα, такими как сарилумаб, может устранить ингибирующее действие ИЛ-6 и восстановить активность цитохрома Р450, приводя к изменению концентрации лекарственных препаратов.
Изменение влияния ИЛ-6 на изоферменты цитохрома Р450 под действием сарилумаба может быть клинически значимо для субстратов цитохрома Р450 с узким терапевтическим диапазоном концентраций, для которых доза корректируется индивидуально. После начала применения или отмены препарата Кевзара® пациентам, получающим лечение лекарственными препаратами, являющимися субстратами цитохрома Р450, следует проводить мониторинг терапевтического эффекта (например, для варфарина) или концентрации лекарственного препарата (например, для теофиллина) и корректировать дозу лекарственного препарата по мере необходимости.
Следует соблюдать осторожность при начале терапии препаратом Кевзара® у пациентов, принимающих препараты, которые являются субстратами изофермента ЗА4 цитохрома Р450 (CYP3A4) (например, оральные контрацептивы или статины), так как сарилумаб может устранить ингибирующий эффект ИЛ-6 и восстанавливать активность изофермента CYP3A4, приводя к снижению экспозиции и активности субстратов CYP3A4. Взаимодействие сарилумаба с субстратами других изоферментов CYP (CYPP2C9, CYP2C19, CYP2D6) не изучалось.
Особые указания
Серьезные инфекции
В период лечения препаратом Кевзара® следует тщательно контролировать пациентов на предмет развития симптомов и признаков инфекций. Поскольку среди пациентов пожилого возраста частота развития инфекций выше, следует с осторожностью проводить лечение данной категории пациентов.
Препарат Кевзара® не следует применять у пациентов с активным течением инфекционного заболевания, включая локализованные инфекции. Необходимо оценить соотношение пользы и риска перед началом применения препарата Кевзара® у пациентов:
- с хронической или рецидивирующей инфекцией;
- с серьезными или оппортунистическими инфекциямив анамнезе;
- с ВИЧ-инфекцией;
- с сопутствующими заболеваниями, предрасполагающими к развитию инфекций;
- после контакта с больными туберкулезом;
- проживавших или посещавших регионы, эндемичные по туберкулезу или микозам.
Следует прервать лечение препаратом Кевзара®, если у пациента отмечается развитие серьезной или оппортунистической инфекции.
Пациент, у которого в период лечения препаратом Кевзара® развилась инфекция, должен незамедлительно пройти полное диагностическое обследование, предусмотренное для лиц с ослабленным иммунитетом; затем ему должна быть назначена адекватная антибактериальная терапия с последующим тщательным наблюдением.
У пациентов, получавших иммунодепрессивные препараты для лечения ревматоидного артрита, включая препарат Кевзара®, были зарегистрированы серьезные инфекции, иногда с летальным исходом, вызванные бактериальными, микобактериальными, инвазивными грибковыми, вирусными и другими оппортунистическими патогенами. Наиболее часто наблюдавшимися серьезными инфекциями при применении препарата Кевзара® были пневмония и целлюлит (воспаление подкожной жировой клетчатки). Из оппортунистических инфекций при применении препарата Кевзара® были зарегистрированы туберкулез, кандидоз и пневмоцистоз. В единичных случаях наблюдались диссеминированные, а не локализованные инфекции у пациентов, часто получающих сопутствующую терапию иммунодепрессивными препаратами, такими как метотрексат или глюкокортикостероиды, что в сочетании с ревматоидным артритом, может предрасполагать к развитию инфекции.
Туберкулез
До начала лечения препаратом Кевзара® у пациентов необходимо оценить наличие факторов риска туберкулеза и провести обследование на латентную инфекцию. Пациентам с латентным или активным туберкулезом следует до начала лечения препаратом Кевзара® провести стандартную противотуберкулезную терапию. У пациентов с латентным или активным туберкулезом в анамнезе, у которых невозможно подтвердить, проводился ли необходимый курс терапии, и у пациентов с отрицательным результатом анализа на латентный туберкулез, но имеющих факторы риска развития туберкулезной инфекции, следует рассмотреть возможность проведения противотуберкулезной терапии до начала лечения препаратом Кевзара®. При решении вопроса о проведении противотуберкулезной терапии целесообразно проконсультироваться с фтизиатром.
Следует тщательно контролировать пациентов на предмет развития признаков и симптомов туберкулеза, включая пациентов, чей результат обследования на латентный туберкулез до начала терапии был отрицательным.
Реактивация вирусной инфекции
При применении иммунодепрессивных биологических препаратов сообщалось о реактивации вирусных инфекций. В клинических исследованиях препарата Кевзара® отмечались случаи опоясывающего герпеса. В клинических исследованиях случаи реактивации вируса гепатита В зарегистрированы не были, однако из исследований были исключены пациенты, имеющие риск реактивации инфекции.
Лабораторные показатели
Количество нейтрофилов
При лечении препаратом Кевзара® отмечалась более высокая частота снижения АЧН. Снижение АЧН не сопровождалось более высокой частотой развития инфекций, включая серьезные инфекции.
Не рекомендуется начинать лечение препаратом Кевзара® у пациентов с АЧН <2×109/л. У пациентов со снижением АЧН <0,5×109/л лечение препаратом Кевзара следует отменить.
Следует контролировать число нейтрофилов через 4-8 недель после начала терапии препаратом Кевзара® и далее – в зависимости от клинических показаний. Рекомендации по изменению дозы на основании значений АЧН представлены в разделе «Способ применения и дозы».
При изменении дозы препарата Кевзара® следует ориентироваться на показатели, полученные в конце интервала между введениями препарата.
Количество тромбоцитов
В клинических исследованиях при лечении препаратом Кевзара® отмечалось снижение количества тромбоцитов. Снижение количества тромбоцитов не сопровождалось развитием кровотечения.
Не рекомендуется начинать лечение препаратом Кевзара® у пациентов с количеством тромбоцитов <150×103/мкл. При снижении количества тромбоцитов <50×103/мкл терапию препаратом Кевзара® следует отменить. Следует контролировать количество тромбоцитов через 4-8 недель после начала терапии и далее – в зависимости от клинических показаний. Рекомендации по изменению дозы препарата на основании количества тромбоцитов представлены в разделе «Способ применения и дозы».
Ферменты печени
При лечении препаратом Кевзара® отмечалась более высокая частота повышения активности «печеночных» ферментов, что в клинических исследованиях носило транзиторный характер и не приводило к появлению каких-либо клинически выраженных симптомов повреждения печени. Увеличение частоты и выраженности повышения активности «печеночных» ферментов наблюдалось при применении препарата Кевзара® в комбинации с потенциально гепатотоксичными препаратами (например, метотрексатом).
Не рекомендуется начинать лечение препаратом Кевзара® у пациентов с повышением активности «печеночных» трансаминаз аланинаминотрансферазы (АЛТ) или аспартатаминотрансферазы (ACT) >1,5×ВГН. При повышении активности АЛТ >5×ВГН терапию препаратом Кевзара® следует отменить.
Следует контролировать активность АЛТ и ACT через 4-8 недель после начала терапии и далее – каждые 3 месяца. В случае клинической необходимости следует рассмотреть возможность исследования других показателей функции печени, таких как билирубин. Рекомендации по изменению дозы на основании повышения активности «печеночных» трансаминаз представлены в разделе «Способ применения и дозы».
Изменение показателей липидного обмена
У пациентов с хроническими воспалительными заболеваниями концентрации липидов в крови могут быть снижены. Лечение препаратом Кевзара® сопровождалось повышением концентраций липидов, таких как холестерин ЛПНП, холестерин ЛПВП и/или триглицериды.
Необходимо контролировать показатели липидного обмена приблизительно через 4-8 недель после начала терапии препаратом Кевзара®, затем – примерно через каждые 6 месяцев.
Лечение пациентов осуществляется согласно клиническим руководствам по ведению пациентов с гиперлипидемией.
Перфорация желудочно-кишечного тракта
В клинических исследованиях было зарегистрировано такое нежелательное явление, как перфорация желудочно-кишечного тракта, которая, прежде всего, является осложнением дивертикулита. Следует с осторожностью применять препарат Кевзара® у пациентов, имеющих в анамнезе указания на язвы желудочно-кишечного тракта или дивертикулит. Необходимо незамедлительно обращать внимание на появление у пациентов новых абдоминальных симптомов, таких как стойкая боль и повышение температуры.
Злокачественные новообразования
Лечение иммунодепрессивными препаратами может приводить к увеличению риска развития злокачественных новообразований. Влияние терапии препаратом Кевзара® на развитие злокачественных новообразований неизвестно, но в клинических исследованиях были зарегистрированы случаи злокачественных новообразований.
Реакции гиперчувствительности
Сообщалось о развитии реакций гиперчувствительности, связанных с приемом препарата Кевзара®. Наиболее частыми реакциями гиперчувствительности были сыпь в месте введения препарата, кожная сыпь и крапивница. Пациента следует информировать о незамедлительном обращении к врачу в случае появления любых реакций гиперчувствительности. При развитии анафилактических реакций или реакций гиперчувствительности введение препарата Кевзара® следует прекратить немедленно. Препарат Кевзара® не назначают пациентам с известной гиперчувствительностью к сарилумабу.
Нарушение функции печени
Пациентам с активными заболеваниями печени или нарушениями функции печени лечение препаратом Кевзара® не рекомендуется.
Вакцинация
Следует избегать одновременного применения живых, а также живых аттенуированных вакцин во время лечения препаратом Кевзара®, поскольку клиническая безопасность данного взаимодействия не установлена. Отсутствуют данные о вторичной передаче возбудителей заболеваний от лиц, вакцинированных живыми вакцинами, пациентам, получающим препарат Кевзара®. Перед началом лечения препаратом Кевзара® рекомендуется, чтобы все пациенты были вакцинированы в соответствии с действующими рекомендациями по вакцинации. Интервал между вакцинацией живыми вакцинами и началом лечения препаратом Кевзара® должен соответствовать действующим рекомендациям по вакцинации в отношении одновременного применения иммунодепрессивных препаратов.
Кардиоваскулярный риск
Пациенты с ревматоидным артритом имеют повышенный риск развития сердечно-сосудистых заболеваний. Факторы риска (например, артериальная гипертензия, гиперлипидемия) необходимо учитывать в рамках проведения стандартной терапии.
Применение у детей
Не следует применять препарат Кевзара® у детей в возрасте до 18 лет (в настоящий момент безопасность и эффективность препарата не установлены).
Несовместимость
Ввиду отсутствия исследований на совместимость, препарат Кевзара® нельзя смешивать с другими лекарственными средствами.
Влияние на способность управлять транспортными средствами и механизмами
Препарат Кевзара® не оказывает или оказывает незначительное влияние на способность управлять транспортными средствами или работать с механизмами.
Форма выпуска
Раствор для подкожного введения 131,6 мг/мл и 175 мг/мл.
По 1,14 мл в одноразовый шприц из прозрачного стекла (тип I), снабженный несъемной иглой из нержавеющей стали, защищенной колпачком из мягкого полимера.
По 2 шприца с инструкцией по применению в картонную пачку с заклеенными клапанами.
По 1 шприцу в шприц-ручку. По 2 шприц-ручки с инструкцией по применению в картонную пачку с заклеенными клапанами.
На каждую картонную пачку нанесен антиконтрафактный стикер.
Условия хранения
При температуре от 2 °С до 8 °С в оригинальной упаковке для защиты от действия света. Не замораживать.
При извлечении из холодильника препарат должен храниться при температуре не выше 25 °С и быть использован в течение 14 дней.
Хранить в недоступном для детей месте.
Срок годности
2 года.
Препарат не следует применять по истечении срока годности.
Условия отпуска
Отпускают по рецепту.
Юридическое лицо, на имя которого выдано регистрационное удостоверение
АО Санофи-авентис груп, Франция.
Производитель
Санофи Винтроп Индустрия, Франция.
Фасовщик (первичная упаковка)
Санофи Винтроп Индустрия, Франция.
Упаковщик (вторичная (потребительская) упаковка)
Санофи Винтроп Индустрия, Франция.
Санофи-Авентис Дойчланд ГмбХ, Германия.
Выпускающий контроль качества
Санофи Винтроп Индустрия, Франция.
Sanofi Winthrop Industrie, France.
1051 boulevard Industriel 76580 Le Trait, France.
Санофи-Авентис Дойчланд ГмбХ, Германия.
Sanofi-Aventis Deutschland GmbH, Germany.
Industriepark Hoechst – Brueningstrasse 50
H500, H590, H600 65926 Frankfurt am Main, Germany.
Претензии потребителей направлять по адресу в России:
АО «Санофи Россия».
125009, г. Москва, ул. Тверская, 22.
В случае использования предварительно заполненной шприц-ручки к инструкции по медицинскому применению лекарственного препарата прилагается «Инструкция по использованию предварительно заполненных одноразовых шприц-ручек»
Инструкция по использованию предварительно заполненных одноразовых шприц-ручек
Кевзара® 150 мг раствор для подкожного введения в предварительно заполненной шприц-ручке
Кевзара® 200 мг раствор для подкожного введения в предварительно заполненной шприц-ручке
На этом рисунке показаны части предварительно заполненной шприц-ручки, содержащей препарат Кевзара®.
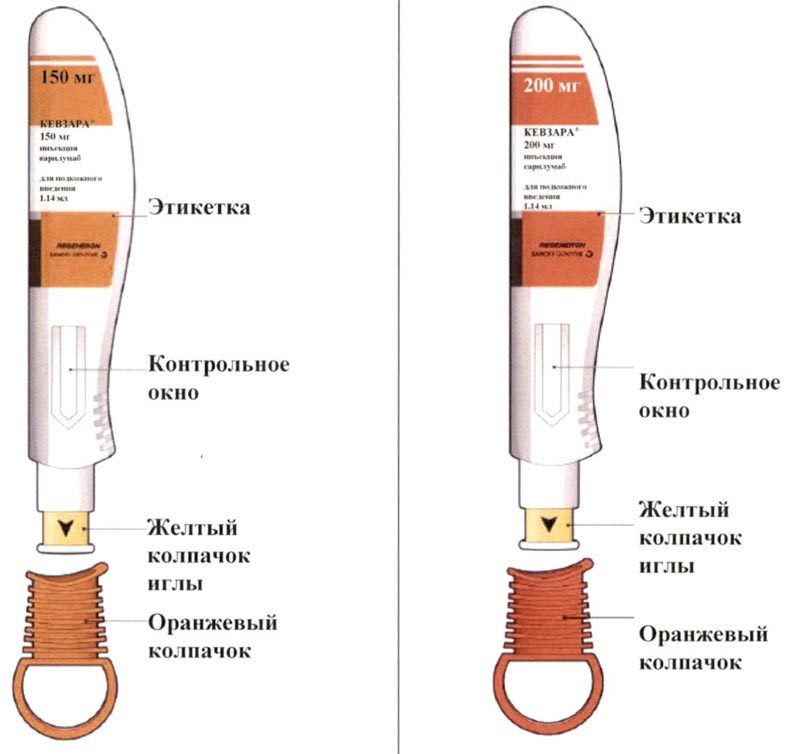
Важная информация
Это устройство представляет собой предварительно заполненную шприц-ручку (в этих инструкциях называемую “шприц-ручка”), содержащую препарат Кевзара® в разовых дозах 150 мг и 200 мг. Препарат вводят подкожно 1 раз каждые 2 недели.
Перед первой инъекцией попросите лечащего врача показать Вам, как правильно использовать шприц-ручку.
Что следует делать
- Перед использованием шприц-ручки внимательно прочитайте все инструкции.
- Убедитесь в том, что Вы используете именно тот лекарственный препарат, который Вам назначил врач, и именно в той дозе, которая Вам рекомендована.
- Неиспользованные шприц-ручки храните в оригинальной картонной пачке в холодильнике при температуре от 2 ° до 8 °С.
- Во время путешествия храните эту картонную пачку в термосумке со льдом.
- Перед использованием оставьте шприц-ручку при комнатной температуре не менее чем на 60 мин ддя того, чтобы она нагрелась.
- Используйте шприц-ручку в течение 14 дней после извлечения ее из холодильника или термосумки.
- Храните шприц-ручку в невидимых и недоступных для детей местах.
Что не следует делать
- Не используйте шприц-ручку, если она повреждена, ее колпачок потерян или не прикреплен.
- Не снимайте колпачок до тех пор, пока Вы не готовы сделать инъекцию.
- Не нажимайте на желтый колпачок иглы и не прикасайтесь к нему пальцами.
- Не пытайтесь надеть колпачок обратно на шприц-ручку.
- Не используйте шприц-ручку повторно.
- Не замораживайте и не нагревайте шприц-ручку.
- Не храните шприц-ручку при температуре выше 25 °С после того, как извлекли ее из холодильника.
- Не подвергайте шприц-ручку воздействию прямых солнечных лучей.
- Не делайте инъекцию через одежду.
Если у Вас имеются любые дополнительные вопросы, обратитесь к лечащему врачу или позвоните по номеру телефона компании, указанному в инструкции.
Шаг А. Подготовьтесь к инъекции.
- На чистой ровной рабочей поверхности разложите все, что Вам понадобится.
- Вам понадобятся спирт, ватный или марлевый тампон, контейнер, резистентный к проколам.
- Возьмите из упаковки одну шприц-ручку, удерживая ее за середину корпуса. Остальные шприц-ручки оставьте в картонной пачке в холодильнике.
- Посмотрите на этикетку.
- Посмотрите на контрольное окно.
- Убедитесь, что жидкость в шприц-ручке прозрачная, от бесцветной до желтоватого цвета.
- Если Вы заметили пузырьки воздуха, это нормальное явление.
- Не вводите препарат, если жидкость мутная, другого цвета или содержит включения.
- Не используйте шприц-ручку, если контрольное окно равномерно желтого цвета.

- Положите шприц-ручку на ровную поверхность и оставьте ее не менее чем на 60 минут для того, чтобы она нагрелась до комнатной температуры (<25 °С).
- Использование шприц-ручки комнатной температуры может сделать инъекцию более комфортной.
- Не используйте шприц-ручку, если она находилась вне холодильника более 14 дней.
- Не нагревайте шприц-ручку; дайте ей нагреться самостоятельно.
- Шприц-ручка не должна подвергаться воздействию прямых солнечных лучей.
- Выберите место инъекции.
- Вы можете сделать инъекцию в наружную поверхность бедра или в переднюю часть живота, за исключением области диаметром 5 см непосредственно вокруг пупка. Если инъекцию препарата делает другой человек, инъекцию также можно сделать в наружную поверхность плеча.
- Каждый раз при введении препарата меняйте место инъекции.
- Не вводите препарат в места с чувствительной, поврежденной кожей или в места с синяками или шрамами.
- Подготовьте место инъекции.
- Вымойте руки.
- Протрите кожу в месте инъекции салфеткой, пропитанной спиртом.
- До введения препарата больше не прикасайтесь к месту инъекции.
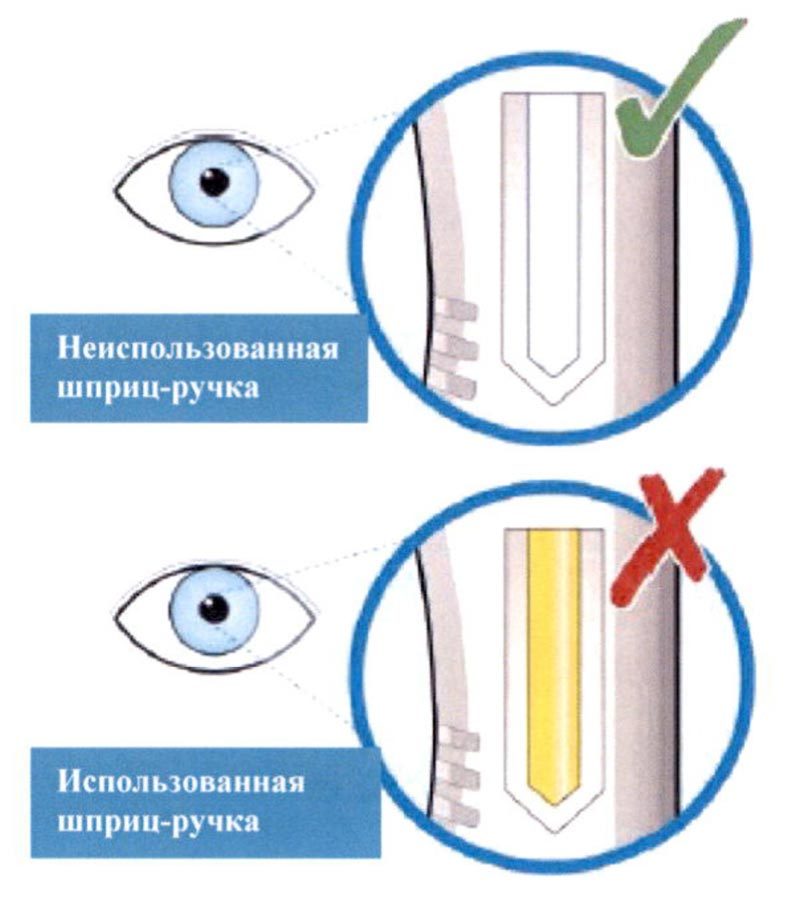
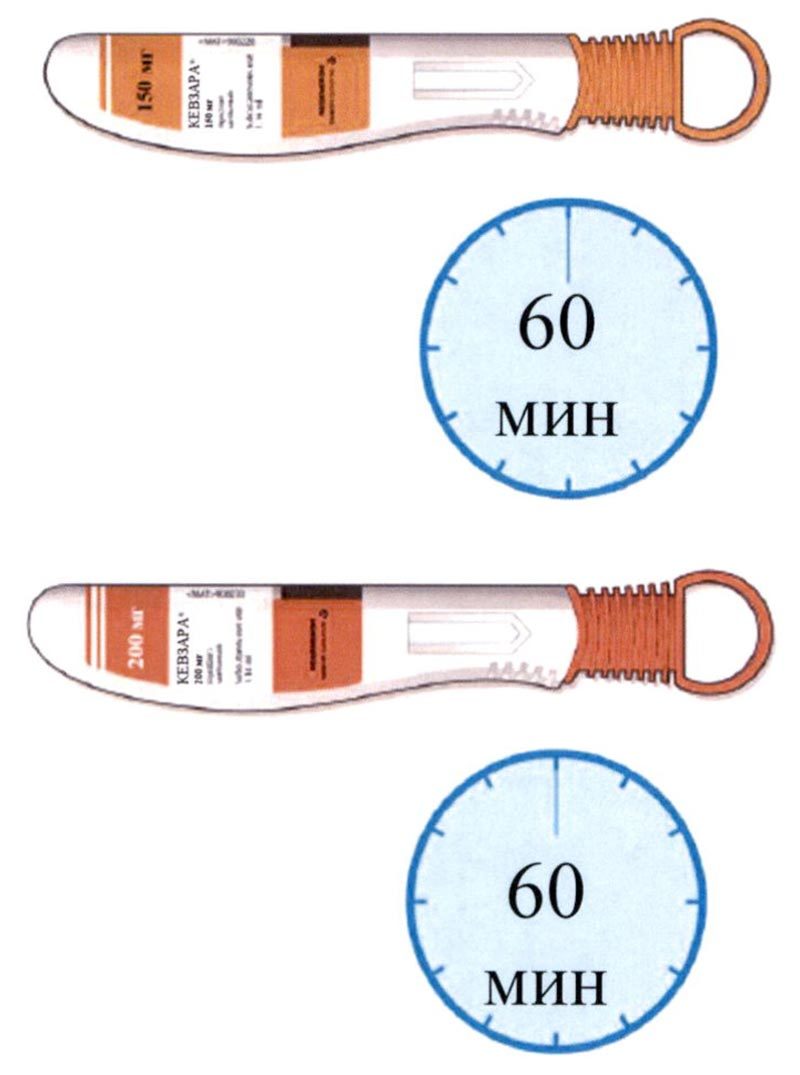
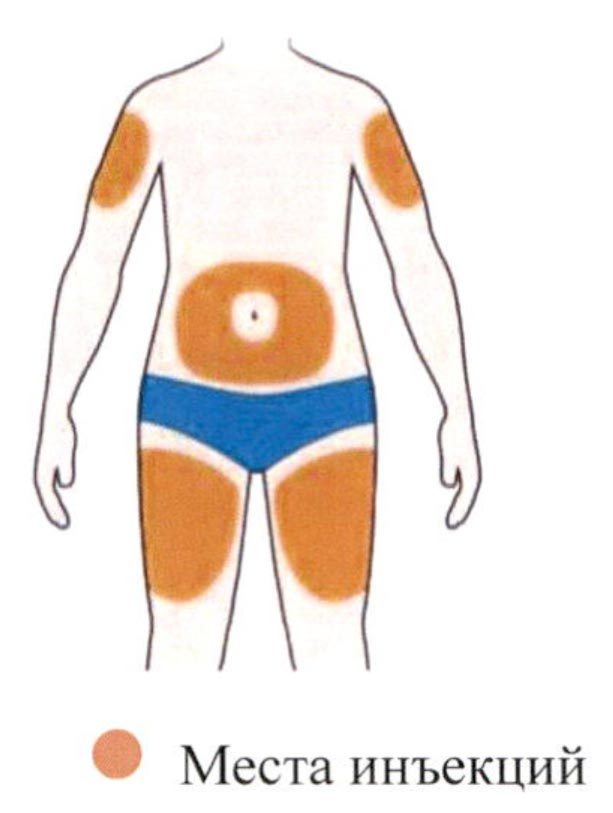
Шаг Б. Выполните инъекцию (Выполняйте Шаг Б только после завершения Шага А «Подготовьтесь к инъекции»)
- Поверните или потяните оранжевый колпачок.
- Не снимайте колпачок с иглы до тех пор, пока не будете готовы выполнить инъекцию.
- Не нажимайте на желтый колпачок иглы и не дотрагивайтесь до него пальцами.
- Не надевайте оранжевый колпачок обратно.
- Прижмите желтый колпачок иглы вертикально к коже примерно под углом 90°.
- Убедитесь, что Вы видите контрольное окно.
- Убедитесь, что Вы видите контрольное окно.
- Нажмите на кожу шприц-ручкой и удерживайте ее.
- Когда начнется введение препарата, Вы услышите щелчок.
- Когда начнется введение препарата, Вы услышите щелчок.
- Продолжайте удерживать шприц-ручку, плотно прижав ее к коже.
- Контрольное окно начнет желтеть.
- Инъекция продолжается до 15 секунд.
- Вы услышите второй щелчок. Прежде чем удалить шприц-ручку, проверьте, стало ли контрольное окно полностью желтым.
- Если Вы не услышали второй щелчок, продолжайте наблюдать за контрольным окном; оно должно стать полностью желтым.
- Если контрольное окно не стало полностью желтым, не вводите вторую дозу без консультации лечащего врача.
- Уберите шприц-ручку от кожи.
- Если Вы заметили кровь, прижмите это место ватным тампоном или марлей.
- Не растирайте кожу после инъекции.
- Положите использованную шприц-ручку и колпачок в контейнер, резистентный к проколам, сразу же после инъекции.
- Всегда храните этот контейнер в невидимых и недоступных для детей местах.
- Не надевайте колпачок обратно.
- Не выбрасывайте использованную шприц-ручку вместе с бытовыми отходами.
- Не применяйте использованный контейнер, резистентный к проколам, для других целей.
- Не выбрасывайте использованный контейнер, резистентный к проколам, вместе с бытовыми отходами, за исключением случаев, когда это разрешается местными правилами. Спросите лечащего врача, как выбрасывать (утилизировать) этот контейнер.
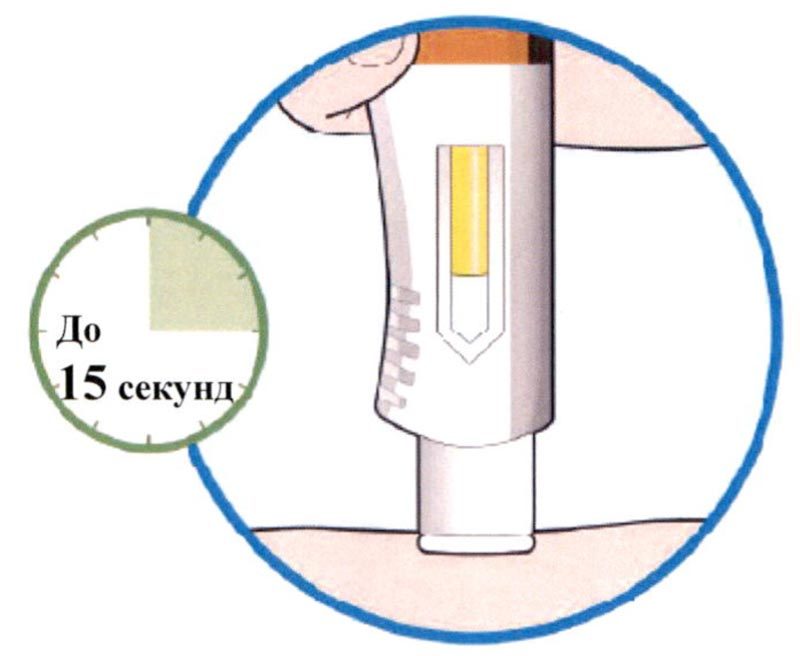
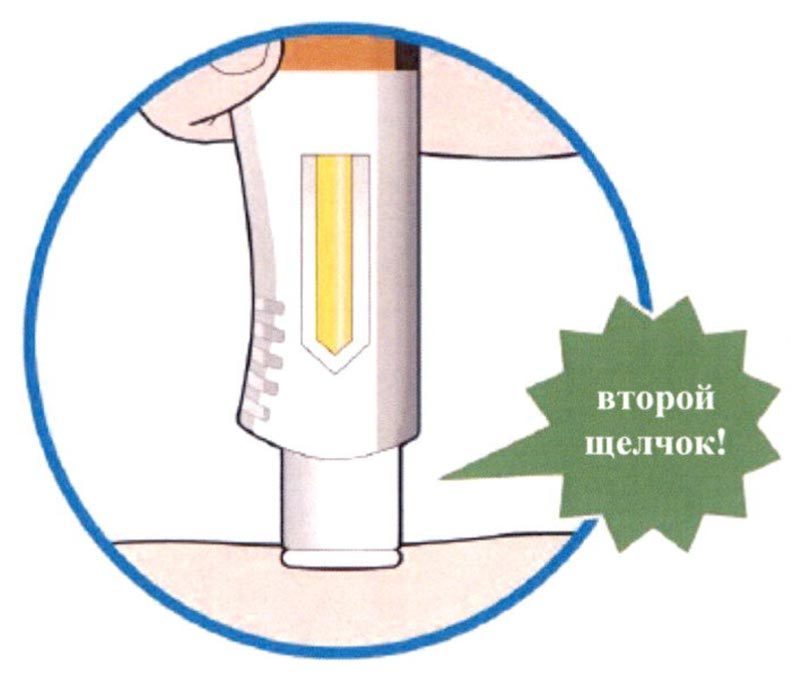
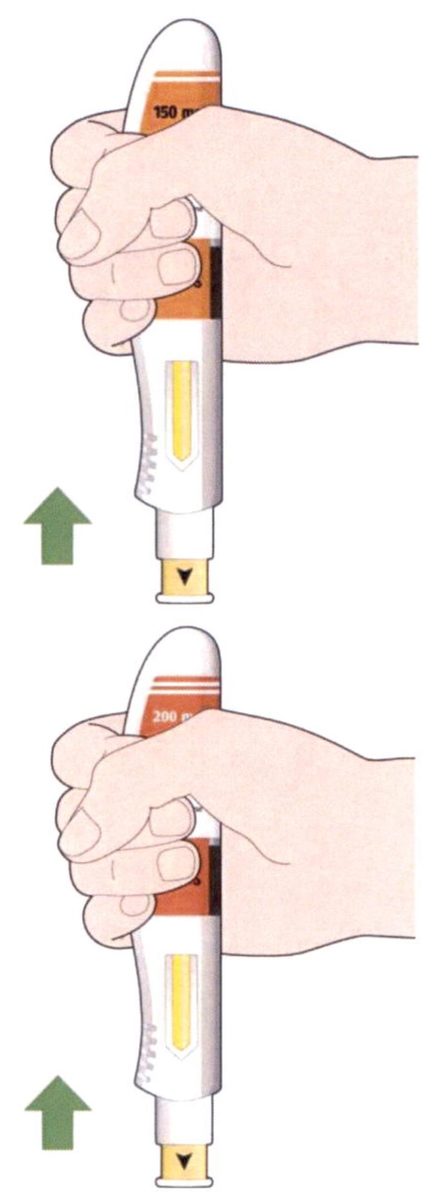
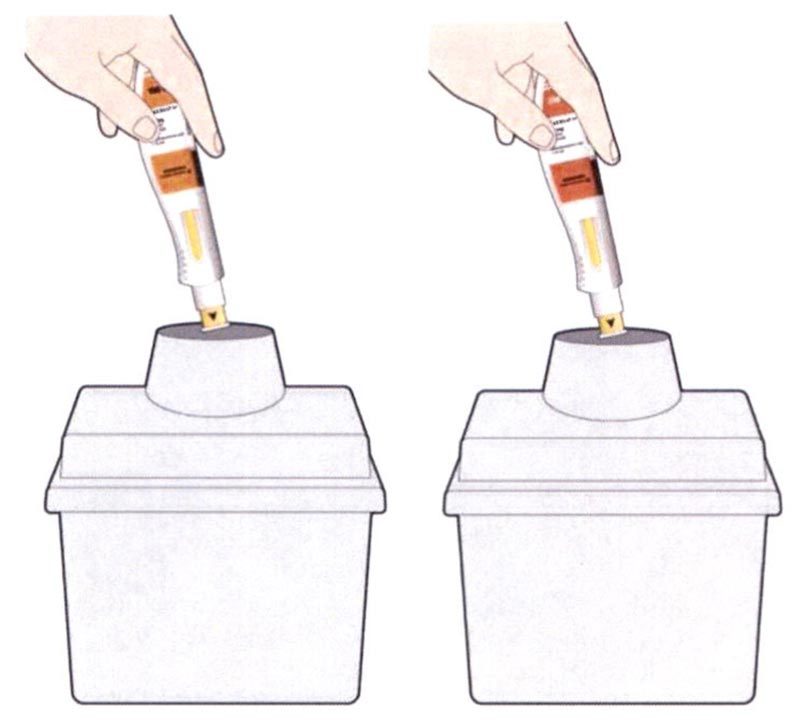
Подготовка и обращение с препаратом
Препараты для парентерального введения должны осматриваться визуально перед введением на предмет наличия видимых частиц и изменения цвета. Если раствор мутный, другого цвета или содержит видимые частицы, его не следует использовать.
После извлечения предварительно заполненной шприц-ручки из холодильника до введения препарата Кевзара® его следует оставить на некоторое время для нагревания до комнатной температуры (< 25 °С).
После извлечения из холодильника препарат Кевзара® следует использовать в течение 14 дней, препарат нельзя хранить при температуре выше 25 °С.
Предварительно заполненные шприц-ручки следует хранить в оригинальной упаковке для защиты от прямых солнечных лучей.
Любое количество неиспользованного препарата или отходы после его использования должны быть утилизированы в соответствии с местными нормативными требованиями.
В случае использования предварительно заполненного шприца к инструкции по медицинскому применению лекарственного препарата прилагается «Инструкция по использованию предварительно заполненных одноразовых шприцев»
Инструкция по использованию предварительно заполненных одноразовых шприцев
Кевзара® 150 мг раствор для подкожного введения в предварительно заполненном шприце
Кевзара® 200 мг раствор для подкожного введения в предварительно заполненном шприце
На этом рисунке показаны части предварительно заполненного шприца, содержащего препарат Кевзара®.
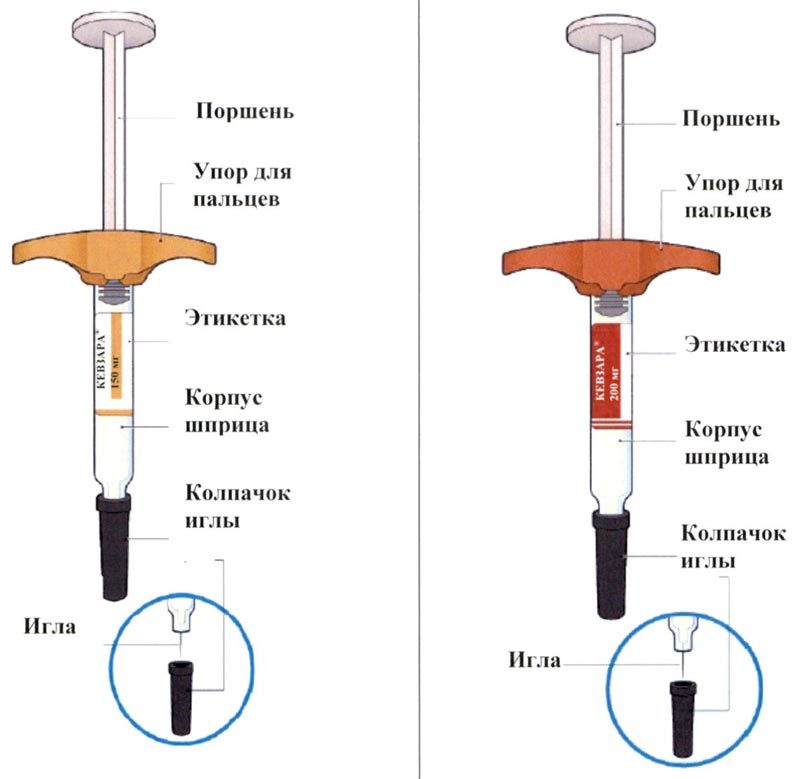
Важная информация
Это устройство представляет собой предварительно заполненный шприц (в этих инструкциях называемый “шприц”), содержащий препарат Кевзара® в разовых дозах 150 мг и 200 мг. Препарат вводят подкожно 1 раз каждые 2 недели.
Перед первой инъекцией попросите лечащего врача показать Вам, как правильно использовать шприц.
Что следует делать
- Перед использованием шприца внимательно прочитайте все инструкции.
- Убедитесь в том, что Вы используете именно тот лекарственный препарат, который Вам назначил врач, и именно в той дозе, которая Вам рекомендована.
- Неиспользованные шприцы храните в оригинальной картонной пачке в холодильнике при температуре от 2 ° до 8 °С.
- Во время путешествия храните эту картонную пачку в термосумке со льдом.
- Перед использованием оставьте шприц не менее чем на 30 мин для того, чтобы он нагрелся до комнатной температуры.
- Используйте шприц в течение 14 дней после извлечения его из холодильника или термосумки.
- Храните шприц в невидимых и недоступных для детей местах.
Что не следует делать
- Не используйте шприц, если он поврежден, или если колпачок иглы отсутствует или не прикреплен.
- Не снимайте колпачок с иглы до тех пор, пока Вы не готовы сделать инъекцию.
- Не прикасайтесь к игле.
- Не пытайтесь надеть колпачок обратно на шприц.
- Не используйте шприц повторно.
- Не замораживайте и не нагревайте шприц.
- Не храните шприц при температуре выше 25 °С после того, как извлекли его из холодильника.
- Не подвергайте шприц воздействию прямых солнечных лучей.
- Не делайте инъекцию через одежду.
Если у Вас имеются любые дополнительные вопросы, обратитесь к лечащему врачу или позвоните по номеру телефона компании, указанному в инструкции.
Шаг А. Подготовьтесь к инъекции.
- На чистой ровной рабочей поверхности разложите все, что Вам понадобится.
- Вам понадобятся спирт, ватный или марлевый тампон, контейнер, резистентный к проколам.
- Возьмите из упаковки один шприц, удерживая его за середину корпуса.
- Остальные шприцы оставьте в картонной пачке в холодильнике.
- Посмотрите на этикетку.
- Проверьте, что у Вас именно тот лекарственный препарат, который Вам назначил врач, и именно в той дозе, которая Вам рекомендована.
- Проверьте срок годности (Годен до).
- Не используйте предварительно заполненный шприц после этой даты.
- Посмотрите на лекарственный препарат.
- Убедитесь, что жидкость в шприце прозрачная, от бесцветной до желтоватого цвета.
- Если Вы заметили пузырьки воздуха, это нормальное явление.
- Не вводите препарат, если жидкость мутная, другого цвета или содержит включения.
- Положите шприц на ровную поверхность и оставьте его не менее чем на 30 мин для того, чтобы он нагрелся до комнатной температуры (<25 °С).
- Использование шприца комнатной температуры может сделать инъекцию более комфортной.
- Не используйте шприц, если он находился вне холодильника более 14 дней.
- Не нагревайте шприц; дайте ему нагреться самостоятельно.
- Шприц не должен подвергаться воздействию прямых солнечных лучей.
- Выберите место инъекции.
- Вы можете сделать инъекцию в наружную поверхность бедра или в переднюю часть живота, за исключением области диаметром 5 см непосредственно вокруг пупка. Если инъекцию препарата делает другой человек, инъекцию также можно сделать в наружную поверхность плеча.
- Каждый раз при введении препарата меняйте место инъекции.
- Не вводите препарат в места с чувствительной, поврежденной кожей или в места с синяками или шрамами.
- Подготовьте место инъекции.
- Вымойте руки.
- Протрите кожу в месте инъекции салфеткой, пропитанной спиртом.
- До введения препарата больше не прикасайтесь к месту инъекции.
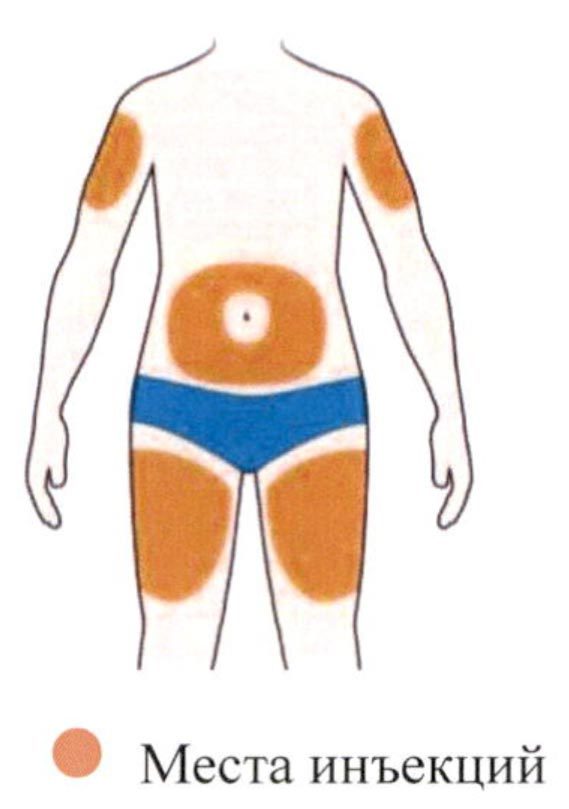
Шаг Б. Выполните инъекцию (выполняйте Шаг Б только после завершения Шага А «Подготовьтесь к инъекции»).
- Снимите колпачок с иглы.
- Держите шприц за середину корпуса так, чтобы игла была направлена в сторону от Вас.
- Не касайтесь поршня руками.
- Не пытайтесь удалить пузырьки воздуха из шприца.
- Не снимайте колпачок с иглы до тех пор, пока не будете готовы выполнить инъекцию.
- Не надевайте колпачок обратно на иглу.
- Зажмите кожу.
- Большим и указательным пальцами слегка зажмите кожу в складку в месте инъекции.
- Большим и указательным пальцами слегка зажмите кожу в складку в месте инъекции.
- Введите иглу в кожную складку под углом 45°.
- Надавите на поршень.
- Медленно надавливайте на поршень до тех пор, пока шприц не будет пустым.
- Медленно надавливайте на поршень до тех пор, пока шприц не будет пустым.
- Перед тем как извлечь иглу убедитесь, что шприц пустой.
- Извлеките иглу под тем же углом, под которым она была введена для выполнения инъекции.
- Если Вы заметили кровь, прижмите это место ватным тампоном или марлей.
- Не растирайте кожу после инъекции.
- Положите использованный шприц и колпачок в контейнер, резистентный к проколам, сразу же после инъекции.
- Всегда храните этот контейнер в невидимых и недоступных для детей местах.
- Не надевайте колпачок обратно на иглу.
- Не выбрасывайте использованный шприц вместе с бытовыми отходами.
- Не применяйте использованный контейнер, резистентный к проколам, для других целей.
- Не выбрасывайте использованный контейнер, резистентный к проколам, вместе с бытовыми отходами, за исключением случаев, когда это разрешается местными правилами. Спросите лечащего врача, как выбрасывать (утилизировать) этот контейнер.
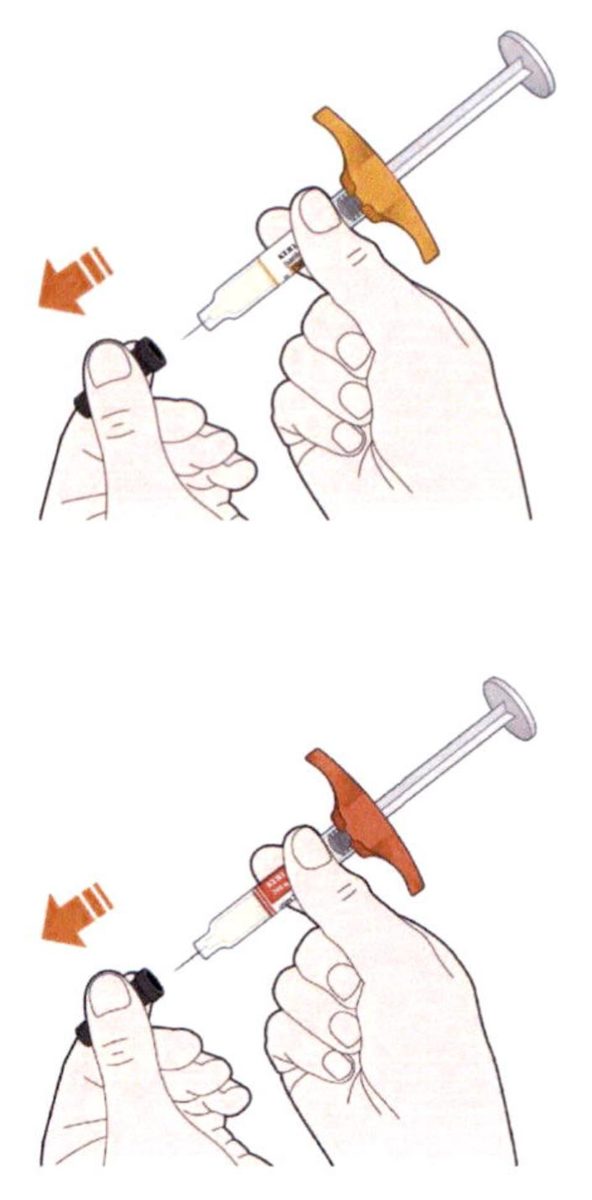
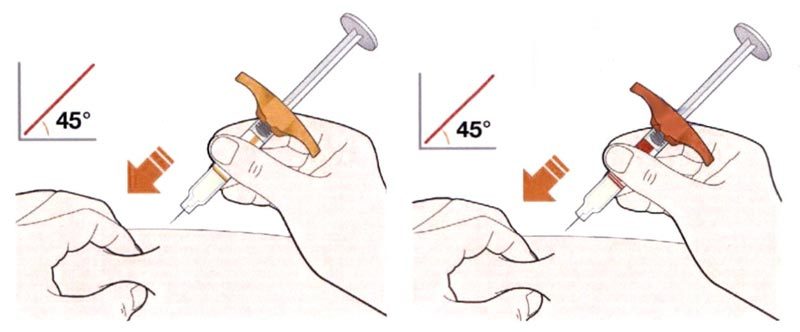
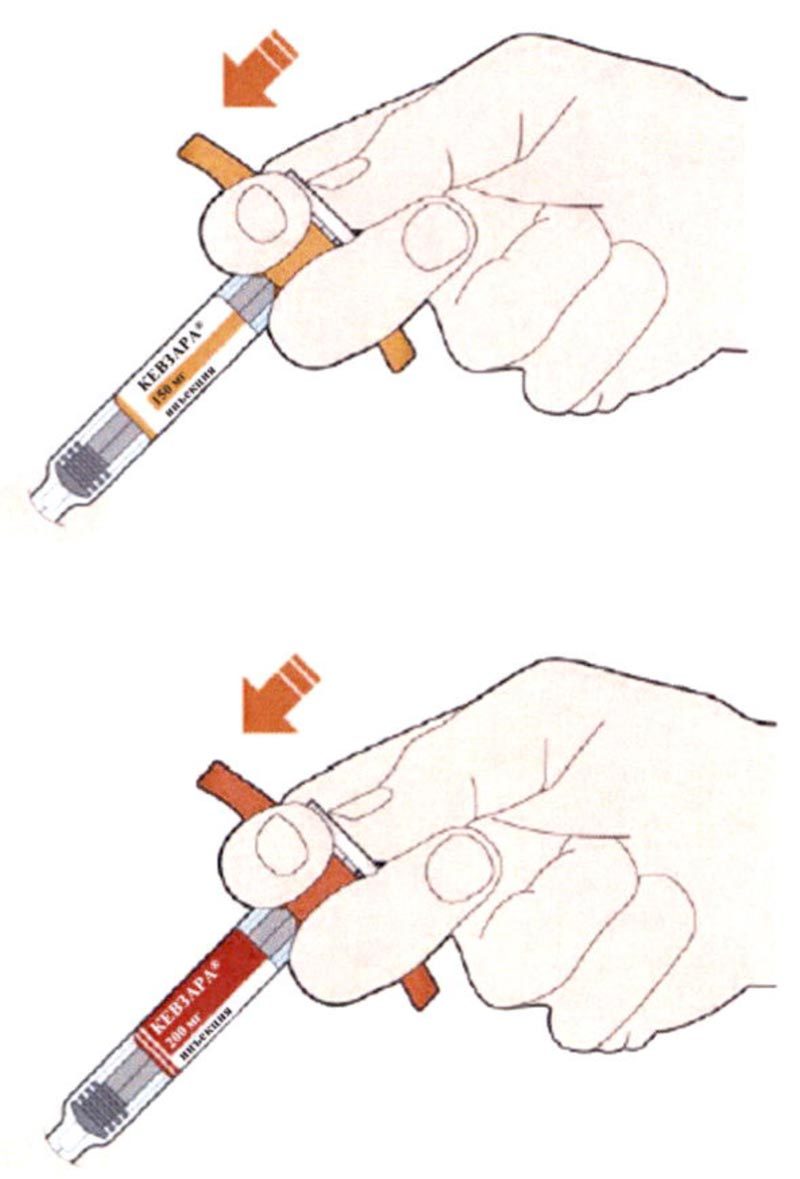
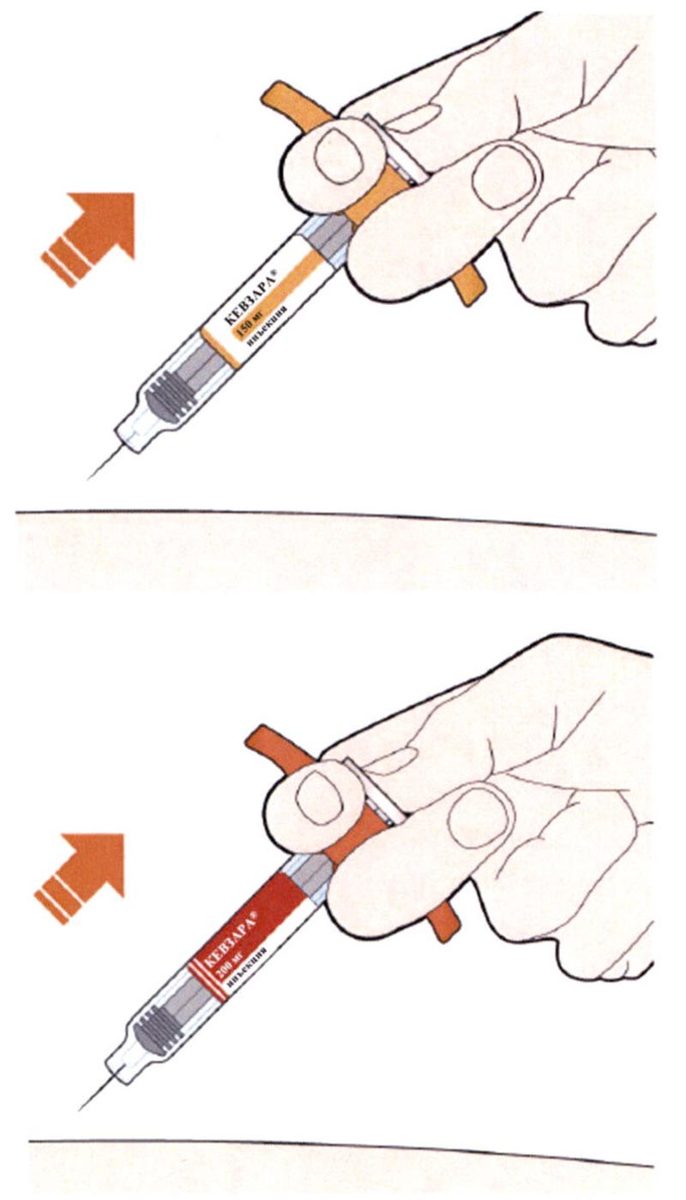
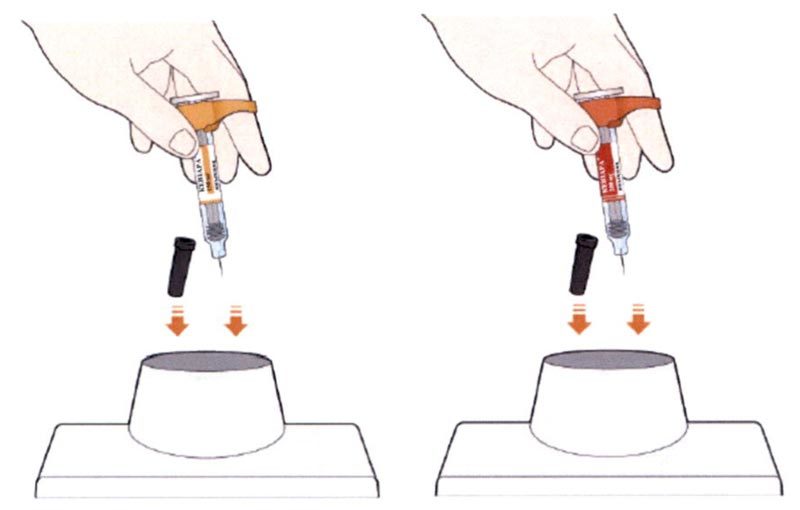
Подготовка и обращение с препаратом
Препараты для парентерального введения должны осматриваться визуально перед введением на предмет наличия видимых частиц и изменения цвета. Если раствор мутный, другого цвета или содержит видимые частицы, его не следует использовать.
После извлечения предварительно заполненного шприца из холодильника до введения препарата Кевзара® его следует оставить на некоторое время для нагревания до комнатной температуры (<25 °С).
После извлечения из холодильника препарат Кевзара следует использовать в течение 14 дней, препарат нельзя хранить при температуре выше 25 °С.
Предварительно заполненные шприцы следует хранить в оригинальной упаковке для защиты от прямых солнечных лучей.
Любое количество неиспользованного препарата или отходы после его использования должны быть утилизированы в соответствии с местными нормативными требованиями.
*Цены в Москве. Точная цена в Вашем городе будет указана на сайте аптеки.
Комментарии
(видны только специалистам, верифицированным редакцией МЕДИ РУ)
Лечение препаратом Кевзара должно назначаться и проводиться под контролем специалистов, имеющих опыт диагностики и лечения ревматоидного артрита.
Рекомендуемая доза препарата Кевзара составляет 200 мг 1 раз каждые 2 недели.
При развитии нейтропении, тромбоцитопении, повышении активности печеночных ферментов рекомендуется уменьшить дозу с 200 мг 1 раз каждые 2 недели до 150 мг 1 раз каждые 2 недели.
Коррекция дозы
При развитии серьезных инфекций следует прекратить лечение препаратом Кевзара до установления контроля над инфекционным процессом.
Не рекомендуется начинать лечение препаратом Кевзара у пациентов со снижением абсолютного числа нейтрофилов (АЧН) менее чем 2х109/л.
Не рекомендуется начинать лечение препаратом Кевзара у пациентов со снижением количества тромбоцитов ниже 150х103/мкл.
Пропуск дозы
Если введение препарата Кевзара пропущено, и с момента пропуска введения препарата прошло 3 дня или менее, то следующую дозу необходимо ввести как можно скорее. Следующую очередную дозу вводят в обычное запланированное время.
Если с момента пропуска введения препарата прошло 4 дня или более, то следующую дозу вводят в следующее обычное запланированное время. При этом дозу нельзя удваивать.
Особые группы пациентов
У пациентов с почечной недостаточностью легкой и средней степени тяжести коррекции дозы препарата не требуется. Применение препарата Кевзара у пациентов с почечной недостаточностью тяжелой степени не изучалось.
Безопасность и эффективность препарата Кевзара не изучались у пациентов с нарушениями функции печени, включая пациентов с положительными результатами серологических исследований на вирус гепатита В (HBV) или вирус гепатита С (HCV).
У пациентов в возрасте старше 65 лет коррекции дозы не требуется.
Не следует применять препарат Кевзара у детей и подростков в возрасте до 18 лет (безопасность и эффективность препарата при ревматоидном артрите не установлены).
Способ применения
Препарат Кевзара вводят п/к.
Все содержимое (1.14 мл) предварительно заполненного шприца/предварительно заполненной шприц-ручки следует вводить п/к.
Места инъекций (область живота, наружная поверхность бедра, наружная поверхность плеча) следует чередовать при каждой инъекции.
Не следует вводить препарат в болезненную и поврежденную кожу, в места с кровоподтеками и рубцами.
Пациент может самостоятельно выполнять п/к инъекцию препарата Кевзара, также инъекцию может выполнять лицо, осуществляющее уход за пациентом. Пациент или лицо, осуществляющее уход за пациентом, до начала применения препарата Кевзара должны быть обучены подготовке и введению препарата.
Инструкция по использованию предварительно заполненных одноразовых шприц-ручек
Устройство представляет собой предварительно заполненную шприц-ручку (в инструкции называемую «шприц-ручка»), содержащую препарат Кевзара в разовых дозах 150 мг и 200 мг. Препарат вводят п/к 1 раз каждые 2 недели.
Перед первой инъекцией следует попросить лечащего врача показать, как правильно использовать шприц-ручку.
Что следует делать:
— перед использованием шприц-ручки внимательно прочитайте все инструкции;
— убедитесь в том, что Вы используете именно тот лекарственный препарат, который Вам назначил врач, и именно в той дозе, которая Вам рекомендована;
— неиспользованные шприц-ручки храните в оригинальной картонной пачке в холодильнике при температуре от +2 до +8 С;
— во время путешествия храните эту картонную пачку в термосумке со льдом;
— перед использованием оставьте шприц-ручку при комнатной температуре не менее чем на 60 мин для того, чтобы она нагрелась;
— используйте шприц-ручку в течение 14 дней после извлечения ее из холодильника или термосумки;
— храните шприц-ручку в невидимых и недоступных для детей местах.
Что не следует делать:
— не используйте шприц-ручку, если она повреждена, ее колпачок потерян или не прикреплен;
— не снимайте колпачок до тех пор, пока Вы не готовы сделать инъекцию;
— не нажимайте на желтый колпачок иглы и не прикасайтесь к нему пальцами;
— не пытайтесь надеть колпачок обратно на шприц-ручку;
— не используйте шприц-ручку повторно;
— не замораживайте и не нагревайте шприц-ручку;
— не храните шприц-ручку при температуре выше +25 С после того, как извлекли ее из холодильника;
— не подвергайте шприц-ручку воздействию прямых солнечных лучей;
— не делайте инъекцию через одежду.
Если у Вас имеются любые дополнительные вопросы, обратитесь к лечащему врачу или позвоните по номеру телефона компании, указанному в инструкции.
Шаг А. Подготовьтесь к инъекции.
1. На чистой ровной рабочей поверхности разложите все, что Вам понадобится.
— спирт, ватный или марлевый тампон, контейнер, резистентный к проколам;
— возьмите из упаковки одну шприц-ручку, удерживая ее за середину корпуса. Остальные шприц-ручки оставьте в картонной пачке в холодильнике.
2. Посмотрите на этикетку.
— проверьте, что у Вас именно тот лекарственный препарат, который Вам назначил врач, и именно в той дозе, которая Вам рекомендована;
— проверьте дату срока годности (Годен до), она указана на боковой стороне шприц-ручки.
Не используйте шприц-ручку после этой даты.
3. Посмотрите на контрольное окно.
— убедитесь, что жидкость в шприц-ручке прозрачная, от бесцветной до желтоватого цвета;
— если Вы заметили пузырьки воздуха, это нормальное явление;
— не вводите препарат, если жидкость мутная, другого цвета или содержит включения;
— не используйте шприц-ручку, если контрольное окно равномерно желтого цвета.
4. Положите шприц-ручку на ровную поверхность и оставьте ее не менее чем на 60 мин для того, чтобы она нагрелась до комнатной температуры (мене +25 С).
— использование шприц-ручки комнатной температуры может сделать инъекцию более комфортной;
— не используйте шприц-ручку, если она находилась вне холодильника более 14 дней;
— не нагревайте шприц-ручку; дайте ей нагреться самостоятельно;
— шприц-ручка не должна подвергаться воздействию прямых солнечных лучей.
5. Выберите место инъекции.
— вы можете сделать инъекцию в наружную поверхность бедра или в переднюю часть живота, за исключением области диаметром 5 см непосредственно вокруг пупка. Если инъекцию препарата делает другой человек, инъекцию также можно сделать в наружную поверхность плеча;
— каждый раз при введении препарата меняйте место инъекции;
— не вводите препарат в места с чувствительной, поврежденной кожей или в места с синяками или шрамами.
6. Подготовьте место инъекции.
— вымойте руки;
— протрите кожу в месте инъекции салфеткой, пропитанной спиртом;
— до введения препарата больше не прикасайтесь к месту инъекции.
Шаг Б. Выполните инъекцию (выполняйте Шаг Б только после завершения Шага А «Подготовьтесь к инъекции»).
1. Поверните или потяните оранжевый колпачок.
— не снимайте колпачок с иглы до тех пор, пока не будете готовы выполнить инъекцию;
— не нажимайте на желтый колпачок иглы и не дотрагивайтесь до него пальцами;
— не надевайте оранжевый колпачок обратно.
2. Прижмите желтый колпачок иглы вертикально к коже примерно под углом 90 градусов.
— убедитесь, что Вы видите контрольное окно.
3. Нажмите на кожу шприц-ручкой и удерживайте ее.
— когда начнется введение препарата, Вы услышите щелчок.
4. Продолжайте удерживать шприц-ручку, плотно прижав ее к коже.
— контрольное окно начнет желтеть;
— инъекция продолжается до 15 сек.
5. Вы услышите второй щелчок. Прежде чем удалить шприц-ручку, проверьте, стало ли контрольное окно полностью желтым.
— если Вы не услышали второй щелчок, продолжайте наблюдать за контрольным окном; оно должно стать полностью желтым;
— если контрольное окно не стало полностью желтым, не вводите вторую дозу без консультации лечащего врача.
6. Уберите шприц-ручку от кожи.
— если Вы заметили кровь, прижмите это место ватным тампоном или марлей;
— не растирайте кожу после инъекции.
7. Положите использованную шприц-ручку и колпачок в контейнер, резистентный к проколам, сразу же после инъекции.
— всегда храните этот контейнер в невидимых и недоступных для детей местах;
— не надевайте колпачок обратно;
— не выбрасывайте использованную шприц-ручку вместе с бытовыми отходами;
— не применяйте использованный контейнер, резистентный к проколам, для других целей;
— не выбрасывайте использованный контейнер, резистентный к проколам, вместе с бытовыми отходами, за исключением случаев, когда это разрешается местными правилами. Спросите лечащего врача, как выбрасывать (утилизировать) этот контейнер.
Инструкция по использованию предварительно заполненных одноразовых шприцев
Устройство представляет собой предварительно заполненный шприц (в инструкции называемый «шприц»), содержащий препарат Кевзара в разовых дозах 150 мг и 200 мг. Препарат вводят п/к 1 раз каждые 2 недели.
Перед первой инъекцией следует попросить лечащего врача показать, как правильно использовать шприц.
Что следует делать
— перед использованием шприца внимательно прочитайте все инструкции;
— убедитесь в том, что Вы используете именно тот лекарственный препарат, который Вам назначил врач, и именно в той дозе, которая Вам рекомендована;
— неиспользованные шприцы храните в оригинальной картонной пачке в холодильнике при температуре от +2 до +8 С;
— во время путешествия храните эту картонную пачку в термосумке со льдом;
— перед использованием оставьте шприц не менее чем на 30 мин для того, чтобы он нагрелся до комнатной температуры;
— используйте шприц в течение 14 дней после извлечения его из холодильника или термосумки;
— храните шприц в невидимых и недоступных для детей местах.
Что не следует делать
— не используйте шприц, если он поврежден, или если колпачок иглы отсутствует или не прикреплен;
— не снимайте колпачок с иглы до тех пор, пока Вы не готовы сделать инъекцию;
— не прикасайтесь к игле;
— не пытайтесь надеть колпачок обратно на шприц;
— не используйте шприц повторно;
— не замораживайте и не нагревайте шприц;
— не храните шприц при температуре выше +25 С после того, как извлекли его из холодильника;
— не подвергайте шприц воздействию прямых солнечных лучей;
— не делайте инъекцию через одежду.
Если у Вас имеются любые дополнительные вопросы, обратитесь к лечащему врачу или позвоните по номеру телефона компании, указанному в инструкции.
Шаг А. Подготовьтесь к инъекции.
1. На чистой ровной рабочей поверхности разложите все, что Вам понадобится.
— спирт, ватный или марлевый тампон, контейнер, резистентный к проколам;
-возьмите из упаковки один шприц, удерживая его за середину корпуса;
— остальные шприцы оставьте в картонной пачке в холодильнике.
2. Посмотрите на этикетку.
— проверьте, что у Вас именно тот лекарственный препарат, который Вам назначил врач, и именно в той дозе, которая Вам рекомендована;
— проверьте срок годности (Годен до);
— не используйте предварительно заполненный шприц после этой даты.
3. Посмотрите на лекарственный препарат.
— убедитесь, что жидкость в шприце прозрачная, от бесцветной до желтоватого цвета;
— если Вы заметили пузырьки воздуха, это нормальное явление;
— не вводите препарат, если жидкость мутная, другого цвета или содержит включения.
4. Положите шприц на ровную поверхность и оставьте его не менее чем на 30 мин для того, чтобы он нагрелся до комнатной температуры (менее +25 С).
— использование шприца комнатной температуры может сделать инъекцию более комфортной;
— не используйте шприц, если он находился вне холодильника более 14 дней;
— не нагревайте шприц; дайте ему нагреться самостоятельно;
— шприц не должен подвергаться воздействию прямых солнечных лучей.
5. Выберите место инъекции.
— вы можете сделать инъекцию в наружную поверхность бедра или в переднюю часть живота, за исключением области диаметром 5 см непосредственно вокруг пупка. Если инъекцию препарата делает другой человек, инъекцию также можно сделать в наружную поверхность плеча;
— каждый раз при введении препарата меняйте место инъекции;
— не вводите препарат в места с чувствительной, поврежденной кожей или в места с синяками или шрамами.
6. Подготовьте место инъекции.
— вымойте руки;
— протрите кожу в месте инъекции салфеткой, пропитанной спиртом;
— до введения препарата больше не прикасайтесь к месту инъекции.
Шаг Б. Выполните инъекцию (выполняйте Шаг Б только после завершения Шага А «Подготовьтесь к инъекции»).
1. Снимите колпачок с иглы.
— держите шприц за середину корпуса так, чтобы игла была направлена в сторону от Вас;
— не касайтесь поршня руками;
— не пытайтесь удалить пузырьки воздуха из шприца;
— не снимайте колпачок с иглы до тех пор, пока не будете готовы выполнить инъекцию;
— не надевайте колпачок обратно на иглу.
2. Зажмите кожу.
— большим и указательным пальцами слегка зажмите кожу в складку в месте инъекции.
3. Введите иглу в кожную складку под углом 45 градусов.
4. Надавите на поршень.
— медленно надавливайте на поршень до тех пор, пока шприц не будет пустым.
Перед тем как извлечь иглу убедитесь, что шприц пустой.
— извлеките иглу под тем же углом, под которым она была введена для выполнения инъекции;
— если Вы заметили кровь, прижмите это место ватным тампоном или марлей;
— не растирайте кожу после инъекции.
6. Положите использованный шприц и колпачок в контейнер, резистентный к проколам, сразу же после инъекции.
— всегда храните этот контейнер в невидимых и недоступных для детей местах;
— не надевайте колпачок обратно на иглу;
— не выбрасывайте использованный шприц вместе с бытовыми отходами;
— не применяйте использованный контейнер, резистентный к проколам, для других целей;
— не выбрасывайте использованный контейнер, резистентный к проколам, вместе с бытовыми отходами, за исключением случаев, когда это разрешается местными правилами. Спросите лечащего врача, как выбрасывать (утилизировать) этот контейнер.
Подготовка и обращение с препаратом
Препараты для парентерального введения должны осматриваться визуально перед введением на предмет наличия видимых частиц и изменения цвета. Если раствор мутный, другого цвета или содержит видимые частицы, его не следует использовать.
После извлечения предварительно заполненной шприц-ручки или предварительно заполненного шприца из холодильника до введения препарата Кевзара их следует оставить на некоторое время для нагревания до комнатной температуры (менее +25 С).
После извлечения из холодильника препарат Кевзара следует использовать в течение 14 дней, препарат нельзя хранить при температуре выше +25 С.
Предварительно заполненные шприц-ручки или предварительно заполненные шприцы следует хранить в оригинальной упаковке для защиты от прямых солнечных лучей.
Любое количество неиспользованного препарата или отходы после его использования должны быть утилизированы в соответствии с местными нормативными требованиями.
Сарилумаб – это новый блокатор рецепторов к интерлейкину-6, который применяется в комбинации с метотрексатом или в виде монотерапии для лечения активного ревматоидного артрита (РА). В двойном слепом, рандомизированном исследовании MOBILITY лечение сарилумабом в сочетании с метотрексатом по сравнению с плацебо привело к уменьшению симптомов, улучшению физической функции и замедлению прогрессирования структурных изменений суставов у больных РА, не отвечавших на метотрексат. В исследовании TARGET эффективность сарилумаба в комбинации с метотрексатом была подтверждена у пациентов с РА, у которых были отменены ингибиторы фактора некроза опухоли α в связи с недостаточной эффективностью или плохой переносимостью. В исследовании MONARCH монотерапия сарилумабом по эффективности превосходила лечение адалимумабом у пациентов с РА, у которых не представлялось возможным применение метотрексата. Профиль безопасности сарилумаба соответствовал ожидаемому профилю безопасности ингибитора интерлейкина-6.
Современная стратегия лечения среднетяжелого и тяжелого ревматоидного артрита (РА) у больных, не отвечающих на метотрексат и другие базисные противовоспалительные препараты (БПВП), предполагает применение генно-инженерных биологических препаратов (ГИБП), прежде всего ингибиторов фактора некроза опухоли-α [1]. Однако в течение года примерно каждый пятый больной прекращает лечение этими препаратами из-за плохой переносимости или неэффективности, а через 3 года доля таких пациентов достигает 50% [2]. В связи с этим на протяжении последних двух десятилетий продолжается разработка новых ГИБП, в частности блокаторов эффектов интерлейкина (ИЛ)-6, гиперпродукция которого играет важную роль в патогенезе РА и многих других иммуновоспалительных заболеваний [3]. ИЛ-6 – это плеотропный провоспалительный цитокин, который вызывает секрецию белков острой фазы гепатоцитами, образование иммуноглобулинов, миграцию и активацию Т- и В-клеток, моноцитов и остеокластов и дает другие эффекты, поддерживающие воспаление и усиливающие деструкцию суставов [4].
Сарилумаб представляет собой человеческое IgG1 моноклональное антитело, которое взаимодействует с растворимыми и мембранными рецепторами ИЛ-6 и блокирует сигнальные системы цитокина [5]. В настоящее время сарилумаб зарегистрирован во многих странах, включая Российскую Федерацию, под торговым наименованием Кевзара® для лечения РА умеренной или высокой степени активности у взрослых пациентов при недостаточной эффективности или непереносимости одного или нескольких БПВП. Выпускается в виде раствора для подкожного введения и применяется в дозе 200 мг каждые 2 недели, которая может быть снижена до 150 мг каждые 2 недели в случае развития нежелательных эффектов. Сарилумаб можно назначать в виде как комбинированной терапии с метотрексатом, так и монотерапии в случае плохой переносимости последнего.
Комбинированная терапия. Эффективность комбинированной терапии сарилумабом и метотрексатом была установлена в многоцентровом, рандомизированном, двойном слепом, плацебо-контролируемом исследовании MOBILITY (часть В), в которое были включены 1197 больных активным РА (счет опухших и болезненных суставов ≥6 и ≥8, соответственно, наличие по крайней мере одной эрозии на рентгенограммах, концентрация С-реактивного белка [СРБ]≥6 мг/л), не отвечавших на метотрексат в стабильной дозе (10-25 мг/нед) в течение по крайней мере 12 недель [6]. Пациенты были рандомизированы на три группы и получали сарилумаб в дозах 150 или 200 мг подкожно каждые 2 недели или плацебо в сочетании с метотрексатом в течение 52 недель. Эффективность лечения оценивали на основании анализа трех первичных конечных точек, включавших частоту ответа по критериям Американской коллегии ревматологов (АКР20) через 24 недели, изменение индекса нетрудоспособности HAQDI через 16 недель и модифицированного счета Шарпа, отражающего выраженность рентгенологических изменений, через 52 недели.
По всем первичным и вторичным показателям эффективности сарилумаб в обеих дозах достоверно превосходил плацебо и вызывал уменьшение симптомов РА, улучшал физическую функцию и задерживал прогрессирование структурных изменений суставов. Эффект препарата проявлялся уже через 2 недели после начала лечения и сохранялся через 52 недели. Например, частота ответа по критериям АКР20 через 24 недели в группах сарилумаба 150 и 200 мг составила 58% и 66%, соответственно, через 52 недели – 54% и 59%, в то время как в группе плацебо в те же сроки на лечение ответили 33% и 32% больных, соответственно (p<0,0001). Частота более выраженного улучшения (АКР50 и АКР70) при лечении сарилумабом также достоверно превышала таковую в группе плацебо (p<0,0001; рис. 1). Ремиссия по критерию DAS28-СРБ (<2,6) через 24 недели была достигнута примерно у трети больных, получавших сарилумаб (27,8-34,1% против 10,1% в группе плацебо; p<0,0001). Около 20% больных, включенных в исследование MOBILITY, имели опыт применения ГИБП в анамнезе. Эффектив ность сарилумаба была сопоставимой у пациентов, получавших и не получавших ГИБП до начала исследования.
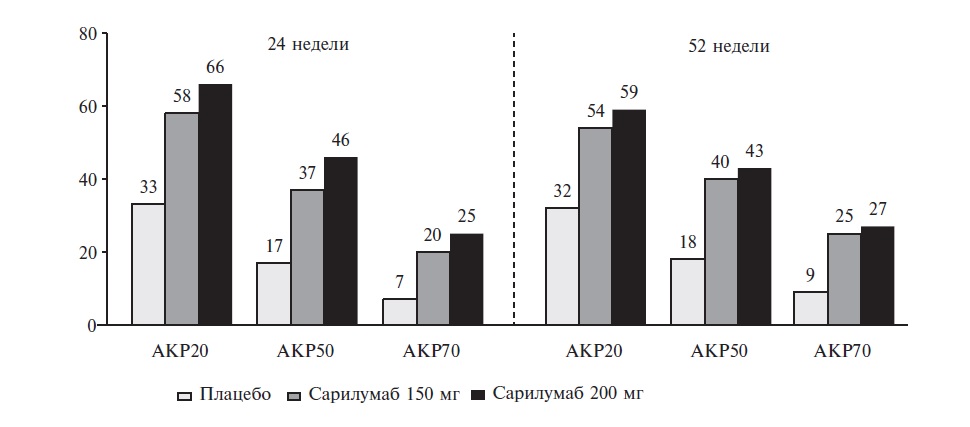
Лечение сарилумабом в дозах 150 и 200 мг по сравнению с плацебо привело к значительному (p<0,0001) улучшению физической функции: через 24 недели счет HAQ-DI в трех группах снизился в среднем на 0,53, 0,55 и 0,29, соответственно. Достигнутый эффект сохранялся через 52 недели. Доля пациентов, ответивших на лечение по этому критерию (снижение счета HAQ-DI по крайней мере на 0,3) в группах сарилумаба была достоверно выше, чем в группе плацебо, как через 24 недели (51,0, 57,4 и 33,4%, соответственно), так и 52 недели (47,0, 47,6 и 26,1%).
Сарилумаб в дозах 150 и 200 мг замедял увеличение модифицированного счета Шарпа (рис. 2), которое отражает прогрессирование рентгенологических изменений в суставах. Причем через 52 недели в группах сарилумаба было выявлено достоверное снижение как счета эрозий, так и счета сужения суставной щели по сравнению с плацебо.
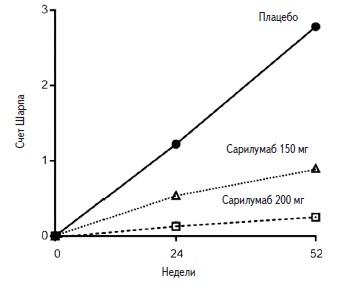
После завершения исследования MOBILITY около 900 пациентов продолжили лечение сарилумабом в дозе 200 мг подкожно каждые 2 недели и метотрексатом открытым методом (исследование EXTEND). Недавно были опубликованы результаты 5-летнего наблюдения этих пациентов, которые показали, что эффект препарата сохраняется при длительном лечении [7]. Например, средний счет DAS28-СРБ в трех пер во начально выделенных группах пациентов был сопо ставимым через 5 лет, причем у больных, рандомизированных в группу сарилумаба 200 мг в двойном слепом исследовании, он снизился на 62% (рис. 3). В выборке ITT частота ремиссии, которую оценивали на основании счета DAS28-СРБ, к концу 5-летнего наблюдения (31-37%) соответствовала таковой в группах сарилумаба в конце исследования MOBILITY. Сходные результаты были получены и при анализе динамики счета CDAI и HAQ. После перевода пациентов с плацебо на сарилумаб было отмечено замедление нарастания счета Шарпа. Тем не менее, через 5 лет показатель прогрессирования структурных изменений суставов в наименьшей степени увеличился в группе больных, которые первоначально получали сарилумаб в дозе 200 мг, а частота отсутствия прогрессирования рентгенологических изменений в группах сарилумаба 150 и 200 мг и плацебо к концу 5-летнего наблюдения составила 42,2, 47,1 и 37,2%, соответственно.
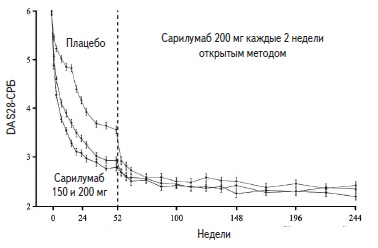
Эффективность сарилумаба в комбинации с метотрексатом у пациентов с РА, прекративших лечение ингибиторами ФНО-α из-за утраты ответа или нежелательных явлений, изучалась в рандомизированном, двойном слепом, плацебо-контролируемом исследовании TARGET [8]. В него были включены 546 больных активным РА, которых рандомизировали на группы сарилумаба 150 и 200 мг и плацебо.
По эффективности, которую оценивали на основании частоты ответа по критериям АКР20 через 24 недели и динамики счета HAQ, сарилумаб в обеих дозах достоверно превосходил плацебо. Частота ответа по АКР20 соответствовала таковой в исследовании MOBILITY и составила 55,8% и 60,9% в группах сарилумаба 150 и 200 мг, соответственно, и 33,7% в группе плацебо (p<0,0001). Частота более выраженного ответа (АКР50 и АКР70) в группах сарилумаба также достоверно превышала таковую в группе плацебо. Ремиссии РА по критерию DAS28-СРБ через 24 недели удалось достичь у 24,9-28,8% больных групп сарилумаба и только у 7,2% пациентов, получавших плацебо. Пре имущества сарилумаба перед плацебо были подтверждены и при анализе динамики счета опухших и болезненных суставов, интенсивности боли в суставах по визуальной аналоговой шкале (ВАШ), общего мнения врачей и пациентов об активности заболевания, концентрации СРБ. Лечение сарилумабом по сравнению с плацебо вызывало также значительное улучшение физической функции (снижение счета HAQ-DI в группах сарилумаба 150 и 200 мг и плацебо составило 0,46, 0,47 и 0,26, соответственно; р<0,001 в обоих случаях).
Монотерапия. Хотя ГИБП пациентам с активным РА рекомендуют назначать в комбинации с метотрексатом, тем не менее, на практике многие пациенты получают монотерапию ГИБП, что может быть связано с плохой переносимостью метотрексата, наличием противопоказаний к его назначению или предпочтениями врачей и пациентов [9,10]. Целью двойного слепого, рандомизированного, контролируемого исследования MONARCH было сравнение эффективности и безопасности монотерапии сарилумабом 200 мг каждые 2 недели или адалимумабом 40 мг каждые 2 недели у 369 больных активным РА, у которых по мнению исследователей было нецелесообразным продолжение приема метотрексата в связи с побочными эффектами или недостаточной эффективностью [11]. Первичным показателем эффективности было снижение счета DAS28-СОЭ через 24 недели. Лечение сарилумабом привело к более значительному снижению этого показателя по сравнению с адалимумабом, причем достоверная разница между группами была достигнута через 12 недель и сохранялась через 24 недели (p<0,0001). Вероятность достижения ремиссии по критерию DAS28-СОЭ при применении сарилумаба по сравнению с адалимумабом была примерно в 3 раза выше через 12 недель (отношение шансов 2,61, 95% доверительный интервал [ДИ] 1,31-5,20; р=0,0051) и в 5 раз выше через 24 недели (4,88, 95% ДИ 2,54-9,39; p<0,0001). Сходные результаты были получены при анализе динамики счета DAS28СРБ. Уже через 4 недели после начала лечения средний счет DAS28-СРБ в группе сарилумаба снизился в большей степени, чем в группе адалимумаба (р=0,0005), а при продолжении терапии ГИБП разница между двумя группами продолжала увеличиваться (рис. 4).
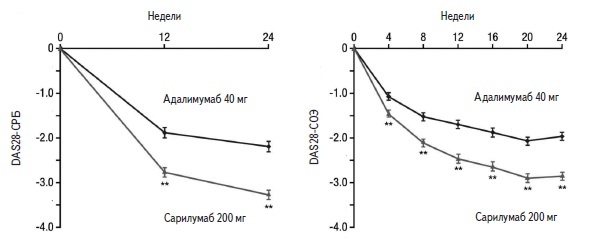
Сарилумаб превосходил также адалимумаб по влиянию на счет CDAI. Величина этого показателя клинического ответа не зависит от концентрации белков острой фазы, динамика которых может быть более выраженной при применении ингибиторов ИЛ-6. У пациентов, получавших сарилумаб, средний счет CDAI через 12 и 24 недели был достоверно ниже, чем у больных группы адалимумаба. Кроме того, через 24 недели в группе сарилумаба доля пациентов, достигших ремиссии или низкой активности РА по критерию CDAI (7,1% и 41,8%, соответственно), была значительно выше, чем в группе адалимумаба (2,7% и 24,9%). В обоих случаях разница была статистически значимой (р=0,0468 и р=0,0005, соответственно).
Через 24 недели в группе сарилумаба было отмечено значительное увеличение частоты ответа по критериям АКР20, АКР50 и АКР70 по сравнению с группой адалимумаба. Разница между группами была достигнута через 8 недель и во всех случаях превышала 10%. Лечение сарилумабом привело также к более выраженному снижению числа опухших и болезненных суставов и улучшению физической функции и показателей качества жизни.
Безопасность
Хотя частота нежелательных явлений при лечении сарилумабом в комбинации с метотрексатом превышала таковую при введении плацебо, тем не менее, переносимость ингибитора ИЛ-6 была в целом хорошей. Например, в двойных слепых, плацебо-контролируемых исследованиях частота серьезных нежелательных явлений у больных, получавших сарилумаб в дозах 150 мг и 200 мг, составила 6,4% и 8,9%, соответственно, а у пациентов группы плацебо – 4,7% [12]. Из-за нежелательных явлений лечение было прекращено у 10,9, 12,6 и 4,7% больных трех групп, соответственно. Наиболее частыми нежелательными эффектами при применении сарилумаба в двух дозах были нейтропения (9,8 и 12,4 против 0,5% в группе плацебо), инфекции верхних дыхательных путей (6,4 и 7,1 против 4,8%), повышение активности АЛТ (6,7 и 6,8 против 2,6%) и эритема в месте введения (5,3 и 5,3 против 0,9%) [12]. При монотерапии сарилумабом чаще всего встречались нейтропения (15,6%), назофарингит (6,0%) и эритема в месте инъекций (6,2%).
Инфекции являются одним из основных нежелательных эффектов любых ГИБП, включая ингибиторы ИЛ-6. Сопутствующая терапия БПВП и/или глюкокортикостероидами может способствовать увеличению риска инфекционных осложнений. В клинических исследованиях частота серьезных инфекций при применении сарилумаба в дозах 150 и 200 мг была сопоставимой с таковой при введении плацебо (3,0, 4,3 и 3,1 на 100 пациенто-лет, соответственно) [13]. Среди серьезных инфекций чаще всего встречались пневмония и целлюлит. В редких случаях отмечалось развитие оппортунистических инфекций, прежде всего опоясывающего герпеса.
Сарилумаб может вызвать снижение числа нейтрофилов и тромбоцитов в крови. В клинических исследованиях в первые 12 недель после начала лечения частота снижения числа нейтрофилов <1 ×109/л при применении сарилумаба в дозах 150 и 200 мг достигала 3,6% и 6,4%, соответственно (против 0% в группе плацебо). Более выраженная нейтропения (<0,5 ×109/л) встречалась значительно реже – у 0,6 и 0,8% пациентов, соответственно [12]. Сниженное число нейтрофилов не сопровождалось увеличением риска развития инфекций и увеличивалось после снижения дозы препарата или перерыва в лечении. Частота тромбоцитопении (<100 × 109/л) при применении сарилумаба в дозах 150 и 200 мг была низкой (0,6 и 1,2%, соответственно). Кроме того, лечение сарилумабом может осложниться увеличением активности аминотрансфераз, которая обычно нормализуется после снижения дозы или временной отмены препарата. В исследовании EXTEND, в которое включали пациентов, завершивших участие в исследовании MOBILITY, снижение дозы сарилумаба с 200 до 150 мг каждые 2 недели было отмечено у 20% из 899 пациентов, продолжавших лечение ингибитором ИЛ-6
Как и при применении другого ингибитора ИЛ-6 – тоцилизумаба, при лечении сарилумабом описаны редкие случаи перфорации желудочно-кишечного тракта, которые в основном были осложнением дивертикулита и чаще наблюдались на фоне сопутствующей терапии нестероидными противовоспалительными препаратами, глюкокортикостероидами или метотрексатом [12].
R. Fleischmann и соавт. недавно обобщили многолетний опыт изучения безопасности сарилумаба у 2887 пациентов, получавших комбинированную терапию, и 471 пациентов, которым проводилась монотерапия этим препаратом [14]. Средняя длительность лечения составила 2,8 и 1,7 года, соответственно, а максимальная – 7,3 и 3,5 года. Самыми частыми нежелательными явлениями при комбинированной терапии были нейтропения, эритема в месте введения и инфекции верхних отделов дыхательных путей, а при монотерапии – нейтропения, эритема в месте введения и назофарингит. Частота серьезных нежелательных явлений при лечении сарилумабом в сочетании с метотрексатом или в виде монотерапии (на 100 пациенто-лет) составила 9,4 и 6,7, соответственно, серьезных инфекций – 3,7 и 1,0, опоясывающего герпеса – 0,6 и 0,5, перфорации желудочно-кишечного тракта – 0,1 и 0, сердечно-сосудистых событий – 0,5 и 0,2, злокачественных опухолей – 0,7 и 0,6. Следует отметить, что частота сердечно-сосудистых исходов и злокачественных опухолей при применении сарилумаба не отличалась от таковой в общей популяции пациентов с РА, а частота серьезных инфекций была несколько ниже, чем при применении тоцилизумаба в сходном анализе [15].
Снижение числа нейтрофилов <1 ×109/л, которое является основанием для перерыва в лечении/снижения дозы сарилумаба, наблюдалось у 13% и 15% больных, получавших комбинированную и монотерапию, соответственно [14]. Частота нейтропении была самой высокой в течение первых 6 мес после начала лечения, а затем снижалась. Нормализация числа нейтрофилов при продолжении комбинированной или монотерапии сарилумабом была отмечена у 70% и 80% пациентов, соответственно. Нейтропения при лечении сарилумабом не сопровождалась развитием серьезных инфекций. Возможно, это связано с тем, что ингибирование ИЛ-6 вызывает миграцию нейтрофилов во внесосудистые депо без ухудшения их функции [16]. В исследованиях in vitro и in vivo блокада ИЛ-6 не оказывала влияния на апоптоз нейтрофильных лейкоцитов, экспрессию молекул адгезии или хемотаксис [17].
Частота увеличения активности АЛТ более чем в 3 раза по сравнению с верхней границей нормы составила 10% и 6% у пациентов, получавших комбинированную и монотерапию сарилумабом, соответственно. Как и частота нейтропении, она была выше в первые 6 мес после начала лечения, а затем снижалась. У большинства больных активность АЛТ нормализовалась при продолжении терапии сарилумабом в сниженной дозе.
В целом исследователи пришли к выводу о том, что профиль безопасности сарилумаба при длительном лечении (до 7 лет) оставался стабильным и соответствовал ожидаемому профилю безопасности ингибитора ИЛ-6 [14].
В исследовании ASCERTAIN были сопоставлены безопасность и переносимость сарилумаба в дозах 150 мг (n=49) и 200 мг (n=51) каждые 2 недели и тоцилизумаба в дозе 4 мг/кг каждые 4 недели, которую при необходимости увеличивали до 8 мг (n=102), у пациентов с РА [18]. Частота нежелательных явлений была сопоставимой в группах сарилумаба и тоцилизумаба. При применении сарилумаба чаще всего встречались нейтропения (12,2-15,7%), назофарингит (5,9-12,2%) и эритема в месте введения (7,8-8,2%), тоцилизумаба – непреднамеренная передозировка (8,8%), инфекции верхних дыхательных путей (6,9%) и тошнота (6,9%). Следует отметить, что средние изменения числа нейтрофилов через 24 недели были сопоставимыми у пациентов, получавших сарилумаб в дозе 200 мг каждые 2 недели, и больных, у которых доза тоцилизумаба была увеличена до 8 мг/кг. По мнению авторов, профиль безопасности двух препаратов существенно не отличался.
Заключение
В двойных слепых, плацебо-контролируемых исследованиях комбинированная терапия сарилумабом и метотрексатом вызывала значительное уменьшение симптомов, улучшала физическую функцию и замедляла прогрессирование структурных изменений суставов у больных активным РА, не отвечавших на метотрексат в адекватной дозе, а также у пациентов, у которых пришлось прекратить терапию ингибиторами ФНО-α в связи с недостаточной эффективностью или плохой переносимостью. Действие сарилумаба проявлялось в течение первых 2 недель, а клинический эффект препарата сохранялся при длительном лечении.
Сарилумаб у пациентов с активным РА может быть использован в виде монотерапии, если применение метотрексата по тем или иным причинам невозможно. В исследовании MONARCH вероятность достижения ремиссии РА при монотерапии сарилумабом была в несколько раз выше, чем при лечении адалимумабом. Это подтверждает тот факт, что при необходимости монотерапии ингибиторы ИЛ-6 могут иметь преимущества по эффективности перед другими ГИБП [1].
В клинических исследованиях профиль безопасности сарилумаба был в целом благоприятным, в частности частота серьезных инфекций оказалась низкой. Лечение сарилумабом, особенно в первые 6 мес, может привести к снижению числа нейтрофилов в крови или повышению активности аминотрансфераз, однако эти лабора торные показатели обычно нормализуются после перерыва в лечении и возобновления терапии сарилумабом в более низкой дозе (150 мг каждые 2 недели).
Судить о сравнительной эффективности сарилумаба и тоцилизумаба не представляется возможным в связи с отсутствием соответствующих исследований. В исследовании ASCERTAIN два ингибитора ИЛ-6 существенно не отличались по профилю безопасности. Сарилумаб при подкожном введении обладает более длительным периодом полувыведения, что позволяет назначать его один раз в 2 недели.
Используемые источники
- Smolen JS, Landewe R, Bijlsma J, et al. EULAR recommendations for the man- agement of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis 2017;76(6):960–77.
- Marchesoni A, Zaccara E, Gorla R, et al. TNF-α antagonist survival rate in a cohort of rheumatoid arthritis patients observed under conditions of standard clin- ical practice. Ann N Y Acad Sci 2009;1173(1):837–46.
- Насонов ЕЛ, Лила АМ. Ингибиция интерлейкина 6 при иммуновоспали- тельных ревматических заболеваниях: достижения, перспективы и надежды. Научно-практическая ревматология 2017;55(6):590-9 [Nasonov EL, Lila AM. Inhibition of interleukin 6 in immune inflammatory rheumatic diseases: achieve- ments, prospects, and hopes. Nauchno-Prakticheskaya Revmatologiya = Rheu — ma tology Science and Practice 2017;55(6):590-9 (In Russ.)].
- Srirangan S, Choy CH. The role of interleukin 6 in the pathophysiology of rheumatoid arthritis. Ther Adv Musculoskel Dis 2010;2(5):247–56.
- Lamb YN, Deeks ED. Sarilumab: A review in moderate to severe rheumatoid arthritis. Drugs 2018;78(9):929-40.
- Genovese MC, Fleischmann R, Kivitz AJ, et al. Sarilumab plus methotrexate in patients with active rheumatoid arthritis and inadequate response to methotrexate: Results of a phase III study. Arthritis Rheumatol 2015;67(6):1424-37.
- Genovese MC, van der Heijde D, Lin Y, et al. Long-term safety and efficacy of sarilumab plus methotrexate on disease activity, physical function and radiograph- ic progression: 5 years of sarilumab plus methotrexate treatment. RMD Open 2019;5(2):e000887.
- Fleischmann R, van Adelsberg J, Lin Y, et al. Sarilumab and nonbiologic disease- modifying antirheumatic drugs in patients with active rheumatoid arthritis and inadequate response or intolerance to tumor necrosis factor inhibitors. Arthritis Rheumatol 2017;69(2):277–90.
- Emery P, Sebba A, Huizinga TW. Biologic and oral disease-modifying anti — rheumatic drug monotherapy in rheumatoid arthritis. Ann Rheum Dis 2013;72: 1897–904.
- Detert J, Klaus P. Biologic monotherapy in the treatment of rheumatoid arthritis. Biologics 2015;9:35–43.
- Burmester GR, Lin Y, Patel R, et al. Efficacy and safety of sarilumab monother- apy versus adalimumab monotherapy for the treatment of patients with active rheumatoid arthritis (MONARCH): a randomised, double-blind, parallel-group phase III trial. Ann Rheum Dis 2017;76(5):840–7.
- European Medicines Agency. Kevzara: CHMP assessment report. 2017. http://www.ema.europa.eu.
- European Medicines Agency. Kevzara: summary of product characteristics 2017. http://www.ema.europa.eu/.
- Fleischmann R, Genovese MC, Lin Y, et al. Long-term safety of sarilumab in rheumatoid arthritis: an integrated analysis with up to 7 years’ follow-up. Rheumatology (Oxford) 2019 Jul 15. pii: kez265. [Epub ahead of print].
- Genovese MC, Rubbert-Roth A, Smolen JS et al. Longterm safety and efficacy of tocilizumab in patients with rheumatoid arthritis: a cumulative analysis of up to 4.6 years of exposure. J Rheumatol 2013;40:768-80.
- Lok LSC, Farahi N, Juss JK et al. Effects of tocilizumab on neutrophil function and kinetics. Eur J Clin Invest 2017;47:736-45.
- Wright HL, Cross AL, Edwards SW, Moots RJ. Effects of IL-6 and IL-6 blockade on neutrophil function in vitro and in vivo. Rheumatology 2014;53:1321-31.
- Emery P, Rondon J, Parrino J, et al. Safety and tolerability of subcutaneous sar- ilumab and intravenous tocilizumab in patients with rheumatoid arthritis. Rheumatology (Oxford) 2019;58(5):849-58.
Версия на английском языке